Atresia de Coanas y Malformación Adenomatoidea Quística.
Revisamos de manera breve las malformaciones congénitas de las vías respiratorias más frecuentes: la atresia de coanas y la malformación congénita de las vías respiratorias inferiores. Entidades raras pero importantes dentro de la patología congénita del recién nacido.
Atresia de Coanas
La atresia de coanas es la obliteración o bloqueo de la abertura nasal posterior. Esta entidad a menudo se asocia con anormalidades óseas de las placas pterigoideas y anormalidades del crecimiento medio facial. Una posible etiología de la atresia de coanas sostiene que la persistencia de la membrana oronasal impide la unión de la nariz y la orofaringe. Esta teoría no tiene en cuenta las anomalías óseas y de la cara media asociadas.
Una teoría alternativa es que las alteraciones en los factores de crecimiento locales resultan en coanas pequeñas o imperforadas. La atresia de coanas ocurre en aproximadamente uno de cada 7000 nacidos vivos. Es más común en las niñas que en los niños. Aproximadamente 2/3 de los casos son unilaterales.
Presentación Clínica de la Atresia de Coanas
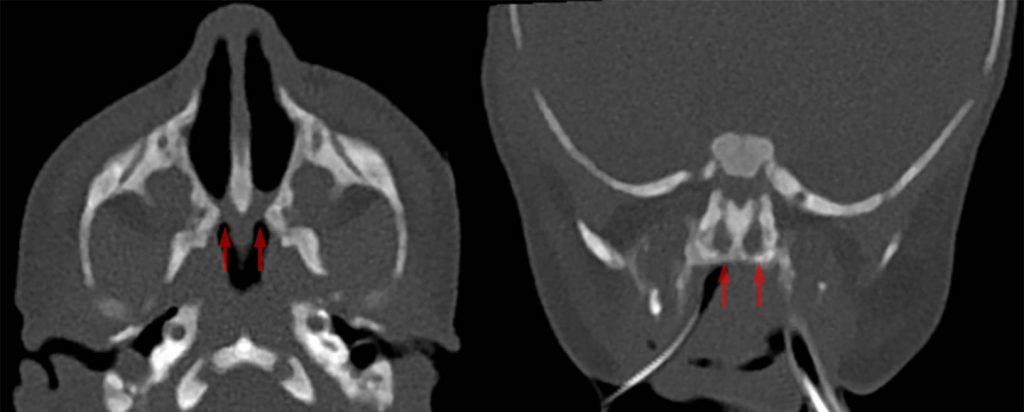
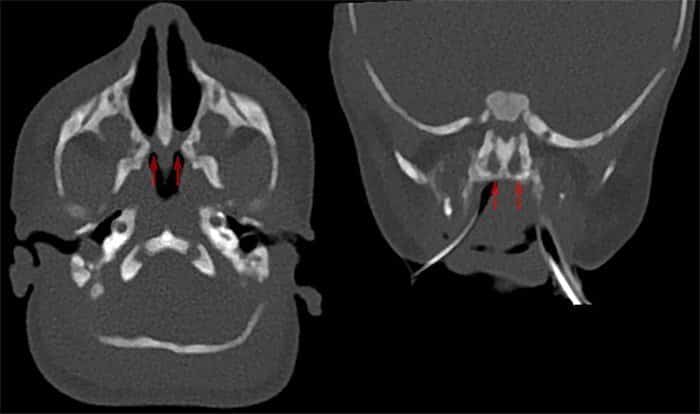
La presentación de la atresia de coanas varía dependiendo de si uno o ambos lados están involucrados. Las personas con atresia de coanas unilateral generalmente se presentan en etapas más tardías con secreción nasal y/u obstrucción unilateral. Los bebés con atresia de coanas bilateral suelen presentar una obstrucción de la vía aérea superior, respiración ruidosa o cianosis que empeora durante la alimentación y mejora cuando el bebé llora. Se debe sospechar el diagnóstico si no se puede pasar un catéter de 5 o 6 French desde la nariz hasta la orofaringe a una distancia de al menos 32 mm.
La medida cualitativa del flujo de aire nasal, como el movimiento de un algodón bajo las fosas nasales o el empañamiento de un espejo, contribuye al diagnóstico clínico. El diagnóstico de atresia de coanas se confirma mediante tomografía computarizada con contraste intranasal que muestra un estrechamiento posterior de la cavidad nasal a nivel de la placa pterigoidea. La atresia de coanas puede ocurrir como una anomalía aislada o como parte de un síndrome de anomalía congénita múltiple.
Por ejemplo, Treacher-Collins, CHARGE (coloboma del iris o coroides, defecto cardíaco, atresia de coanas, retraso en el crecimiento y desarrollo, anormalidades genitourinarias y defectos del oído con sordera asociada), Kallmann, asociación VACTERL/VATER (anomalías vertebrales, atresia anal, defectos cardíacos, fístula traqueoesofágica y/o atresia esofágica, anomalías renales y radiales, y defectos de las extremidades), así como Pfeiffer. Otras anomalías congénitas están presentes en 50 y 60% de los individuos con atresia de coanas unilateral y bilateral, respectivamente.
Además de un examen físico completo para detectar anomalías asociadas, se debe interconsultar a cardiología y oftalmología en los bebés con atresia de coanas. Las anomalías asociadas incluyen:
- Deformidades faciales, nasales o palatinas.
- Polidactilismo
- Cardiopatía congénita
- Coloboma del iris y retina.
- Discapacidad intelectual
- Malformaciones del oído externo.
- Atresia esofágica
- Craneosinostosis
- Fístula traqueoesofágica.
- Meningocele
Manejo de la Atresia de Coanas
El manejo inmediato de los lactantes con atresia de coanas incluye la colocación de una vía aérea oral y el inicio de la alimentación por sonda. La reparación definitiva implica la punción transnasal y la colocación de stent o la resección endoscópica del tabique nasal posterior mediante un abordaje transnasal con o sin colocación de stent. La punción transnasal ha perdido popularidad debido a una tasa de recurrencia inaceptable.
Las ventajas del abordaje endoscópico transnasal incluyen una visión clara del campo operatorio y la eliminación precisa de la placa atrésica y vómer posterior sin dañar las estructuras circundantes. El enfoque transpalatal clásico está reservado para casos difíciles o recurrentes. La recurrencia de la estenosis puede ocurrir incluso después de una cirugía exitosa.
Malformación Congénita de las Vías Respiratorias Inferiores
La malformación congénita de las vías respiratorias inferiores, anteriormente conocida como malformación adenomatoide quística congénita, es una anomalía del desarrollo del tracto respiratorio. Las malformación congénita de las vías respiratorias inferiores se clasifica en los tipos 0 a 4, que se definen patológicamente y tienen diferentes presentaciones clínicas y pronósticos.
La malformación congénita de las vías respiratorias inferiores tipo 1 comprende aproximadamente el 70% de los casos. Se pueden detectar grandes quistes en la ecografía prenatal y pueden causar dificultad respiratoria en el neonato. Los quistes más pequeños pueden presentarse meses o años más tarde como hallazgos incidentales en estudios de imagen o como foco de infección pulmonar recurrente. La malformación congénita de las vías respiratorias inferiores tipo 1 se asocia con un ligero riesgo de malignidad. Se debe tener cuidado de no clasificar erróneamente las lesiones tipo 4 como tipo 1, ya que las lesiones tipo 4 tienen un alto potencial de malignidad.
Las malformación congénita de las vías respiratorias inferiores de tipo 4 comprende del 5 al 10% de todos los casos. Muchas de estas lesiones son formas quísticas de blastoma pleuropulmonar (PPB), por lo que la identificación y resección de estas lesiones es esencial. Las claves importantes que sugieren este tipo de malformación congénita de las vías respiratorias inferiores son la presentación con neumotórax, la presencia de quistes pulmonares bilaterales o multifocales, o antecedente familiar de quistes pulmonares o afecciones asociadas con PPB.
Diagnóstico por Imagen
Todos los bebés con un diagnóstico prenatal de malformación congénita de las vías respiratorias inferiores deben someterse a una radiografía de tórax en el período neonatal, incluso si están asintomáticos e incluso si la lesión parece resolverse en ecografías prenatales seriadas. Además, a todos los bebés se les debe realizar una tomografía computarizada (TAC) o imágenes de resonancia magnética (RM). El momento ideal para la toma de los estudios depende de los síntomas y de los resultados de la radiografía de tórax inicial. La TAC o la RM se deben realizar incluso en bebés con radiografías de tórax normales porque las radiografías simples a menudo no detectan esta entidad en neonatos asintomáticos.
Manejo de la Malformación congénita de las vías respiratorias inferiores
En pacientes con malformación congénita de las vías respiratorias inferiores que está causando algún síntoma respiratorio (dificultad respiratoria o taquipnea), se recomienda resección quirúrgica en lugar de observación. Todos los quistes resecados deben examinarse cuidadosamente para detectar datos de malignidad. Los lactantes asintomáticos con un diagnóstico prenatal de malformación congénita de las vías respiratorias inferiores deben evaluarse con una radiografía de tórax en el período neonatal. El manejo posterior depende de si hay características que sugieran un alto riesgo de complicaciones
Para lactantes y niños con características de alto riesgo (lesiones grandes en la radiografía de tórax, quistes bilaterales o multifocales, antecedentes familiares de afecciones asociadas a blastoma pleuropulmonar o neumotórax), se recomienda la resección quirúrgica temprana en lugar de observación. Estos bebés deben ser evaluados con tomografía computarizada o resonancia magnética antes de la cirugía para confirmar el diagnóstico y seguir evaluando la lesión. Para bebés y niños con lesiones pequeñas y ninguna de las otras características de alto riesgo descritas anteriormente, la resección quirúrgica electiva o el tratamiento conservador con observación son opciones razonables. Si se elige la resección quirúrgica para estos pacientes, generalmente se realiza después del período neonatal pero antes de los 12 meses de edad.
Si se elige la observación, se recomienda un seguimiento clínico cercano durante el primer año de vida para monitorear el desarrollo de síntomas de dificultad respiratoria o infección, así como realizar imágenes de TAC o RM a los seis meses de edad y anualmente a partir de entonces. Para los bebés asintomáticos al nacimiento, el riesgo de desarrollar una infección no está bien delineado. Está claro que algunos de estos bebés desarrollarán infección durante los primeros años de vida si no se realiza la cirugía, pero las estimaciones del riesgo de infección varían de 3 a 30%. Para la mayoría de los lactantes con malformación congénita de las vías respiratorias inferiores, la resección quirúrgica en el período neonatal es curativa y el pronóstico excelente.
Referencias Bibliográficas
Keller JL, Kacker A. Choanal atresia, CHARGE association, and congenital nasal stenosis. Otolaryngol Clin North Am 2000; 33:1343.
Hengerer AS, Brickman TM, Jeyakumar A. Choanal atresia: embryologic analysis and evolution of treatment, a 30-year experience. Laryngoscope 2008; 118:862.
Burrow TA, Saal HM, de Alarcon A, et al. Characterization of congenital anomalies in individuals with choanal atresia. Arch Otolaryngol Head Neck Surg 2009; 135:543.
Friedman NR, Mitchell RB, Bailey CM, et al. Management and outcome of choanal atresia correction. Int J Pediatr Otorhinolaryngol 2000; 52:45.
Cedin AC, Atallah AN, Andriolo RB, et al. Surgery for congenital choanal atresia. Cochrane Database Syst Rev 2012; :CD008993.
Eladl HM, Khafagy YW. Endoscopic bilateral congenital choanal atresia repair of 112 cases, evolving concept and technical experience. Int J Pediatr Otorhinolaryngol 2016; 85:40.
Baird R, Puligandla PS, Laberge JM. Congenital lung malformations: informing best practice. Semin Pediatr Surg 2014; 23:270.
Taguchi T, Suita S, Yamanouchi T, et al. Antenatal diagnosis and surgical management of congenital cystic adenomatoid malformation of the lung. Fetal Diagn Ther 1995; 10:400.
Gajewska-Knapik K, Impey L. Congenital lung lesions: Prenatal diagnosis and intervention. Semin Pediatr Surg 2015; 24:156.
Priest JR, Williams GM, Hill DA, et al. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol 2009; 44:14.
Gornall AS, Budd JL, Draper ES, et al. Congenital cystic adenomatoid malformation: accuracy of prenatal diagnosis, prevalence and outcome in a general population. Prenat Diagn 2003; 23:997.
De Santis M, Masini L, Noia G, et al. Congenital cystic adenomatoid malformation of the lung: antenatal ultrasound findings and fetal-neonatal outcome. Fifteen years of experience. Fetal Diagn Ther 2000; 15:246.
van Koningsbruggen S, Ahrens F, Brockmann M, et al. Congenital cystic adenomatoid malformation type 4. Pediatr Pulmonol 2001; 32:471.
Stocker JT. Cystic lung disease in infants and children. Fetal Pediatr Pathol 2009; 28:155.
De Felice C, Di Maggio G, Messina M, et al. Congenital cystic adenomatoid malformation of the lung associated with esophageal atresia and tracheoesophageal fistula. Pediatr Surg Int 1999; 15:260.
Krous HF, Harper PE, Perlman M. Congenital cystic adenomatoid malformation in bilateral renal agenesis. Its mitigation of Potter’s syndrome. Arch Pathol Lab Med 1980; 104:368.
Chowdhury MM, Chakraborty S. Imaging of congenital lung malformations. Semin Pediatr Surg 2015; 24:168.
Kim WS, Lee KS, Kim IO, et al. Congenital cystic adenomatoid malformation of the lung: CT-pathologic correlation. AJR Am J Roentgenol 1997; 168:47.
Cass DL, Crombleholme TM, Howell LJ, et al. Cystic lung lesions with systemic arterial blood supply: a hybrid of congenital cystic adenomatoid malformation and bronchopulmonary sequestration. J Pediatr Surg 1997; 32:986.
Conran RM, Stocker JT. Extralobar sequestration with frequently associated congenital cystic adenomatoid malformation, type 2: report of 50 cases. Pediatr Dev Pathol 1999; 2:454.
Schwartz MZ, Ramachandran P. Congenital malformations of the lung and mediastinum–a quarter century of experience from a single institution. J Pediatr Surg 1997; 32:44.
Gulack BC, Leraas HJ, Ezekian B, et al. Outcomes following elective resection of congenital pulmonary airway malformations are equivalent after 3 months of age and a weight of 5 kg. J Pediatr Surg 2017.
Khalek N, Johnson MP. Management of prenatally diagnosed lung lesions. Semin Pediatr Surg 2013; 22:24.
Truitt AK, Carr SR, Cassese J, et al. Perinatal management of congenital cystic lung lesions in the age of minimally invasive surgery. J Pediatr Surg 2006; 41:893.
Albanese CT, Sydorak RM, Tsao K, Lee H. Thoracoscopic lobectomy for prenatally diagnosed lung lesions. J Pediatr Surg 2003; 38:553.
Feinberg A, Hall NJ, Williams GM, et al. Can congenital pulmonary airway malformation be distinguished from Type I pleuropulmonary blastoma based on clinical and radiological features? J Pediatr Surg 2016; 51:33.
MacSweeney F, Papagiannopoulos K, Goldstraw P, et al. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol 2003; 27:1139.
Federici S, Domenichelli V, Tani G, et al. Pleuropulmonary blastoma in congenital cystic adenomatoid malformation: report of a case. Eur J Pediatr Surg 2001; 11:196.
Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg 2003; 12:17.
Dusmet M. Adult lung tumours of childhood origin. Semin Pediatr Surg 2015; 24:196.




