Mieloma Múltiple: Abordaje Diagnóstico para una Atención Temprana.

El mieloma múltiple representa aproximadamente el 10 % de las neoplasias malignas hematológicas. Te mencionamos lo más relevante para su diagnóstico para una atención temprana.
Se trata de una gammapatía monoclonal caracterizada por la proliferación tumoral de las células plasmáticas que se acumulan en la médula ósea y como consecuencia inducen la aparición de lesiones osteolíticas y bandas monoclonales. De acuerdo con la Organización Mundial de la Salud (OMS), el mieloma múltiple representa 1% de todas las neoplasias malignas y 10-15% de las neoplasias hematológicas. Es una enfermedad heterogénea en el año 2015 fueron diagnosticadas 124.225 personas y se estima que esa cifra ascenderá a cerca del 30 por ciento para el 2025.
El 90% de los casos de mieloma múltiple son pacientes mayores de 50 años. El riesgo de padecer mieloma múltiple es 3.7 veces mayor para los individuos con un familiar de primer grado que haya sufrido la enfermedad.
Genética en el Mieloma Múltiple
Se piensa que el mieloma evoluciona más comúnmente desde una gammapatía monoclonal de importancia conocida como MGUS que progresa hasta el mieloma latente. Varias anomalías genéticas que ocurren en las células plasmáticas tumorales juegan un papel importante en la patogénesis del mieloma. Las translocaciones cromosómicas tempranas primarias ocurren en la revisión del interruptor de la inmunoglobulina en el cromosoma 14q32,33.
Las translocaciones secundarias de inicio tardío y la simulaciones genéticas implicadas en la progresión de la enfermedad incluyen anomalías cariotípicas complejas en MYC activación de NRAS y KRAS, mutaciones en FGFR3 (receptor 3 del factor de crecimiento de fibroblastos) y TP53 (gen p53), y la inactivación de inhibidores de quinasa dependientes de cíclica CDKN2A y CDKN2C.
Fisiopatología del Mieloma Múltiple
Hay varios tipos de neoplasias de células plasmáticas. Todas estas enfermedades se relacionan con una proteína (proteína M) monoclonal (o mieloma). Ellas incluyen la gammapatía monoclonal de significación indeterminada (GMSI), el plasmocitoma solitario de hueso, el plasmocitoma extramedular y el mieloma múltiple. La adición de células de mieloma a células hematopoyéticas y estromales induce la secreción de citoquinas y factor de crecimiento endotelial vascular (VEGF) factor de crecimiento similar a la insulina 1 e IL-10.
Estas citoquinas y factores de crecimiento son producidos y secretados por células en la médula ósea. La adhesión de células de mieloma a proteínas de matriz extracelular desencadena la regulación de proteínas reguladoras del ciclo celular y de proteínas antiapoptóticas. Las lesiones óseas son causadas por un desequilibrio en la función de los osteoblastos y osteoclastos. La inhibición de la vía Wnt suprime a los osteoblastos, mientras que la amplificación de la vía RANK y la acciónn de la proteína inflamatoria macrófaga 1 alfa (MIP1α) activan los osteoclastos.
Las células de mieloma múltiple alteran la homeostasis de las células estromales y la interacción entre ésta. La matriz extracelular, citocinas y factores de crecimiento; como consecuencia las células tumorales inducen secuelas de señalización directas e indirectas en la médula ósea. Estas promueven la proliferación, supervivencia, migración y resistencia a medicamentos de las células de mieloma múltiple. La enfermedad ósea observada en mieloma múltiple. La enfermedad ósea observada en mieloma múltiple, presente en 80% de los pacientes, refleja el desequilibrio existe entre los osteoclastos y osteoblastos.
Presentación Clínica del Mieloma Múltiple
Se distingue por dolor óseo grave (25%), fracturas patológicas vertebrales (30%) y extravertebrales e hipercalcemia (12%). Estos eventos esqueléticos no sólo ejercen un efecto negativo en la calidad de vida de los pacientes, sino también disminuyen su tiempo de supervivencia. La manifestación clínica más frecuente es el síndrome anémico. Hasta 73% de los pacientes con mieloma múltiple presentan concentraciones de hemoglobina menores de 12 g/dl y un 82% presenta fatiga.
La hipercalcemia se identifica en 18-30% de los casos al momento del diagnóstico inicial. Alrededor de 13% tiene concentraciones de calcio mayores de 11 mg/dl. La hiperviscosidad sanguínea suele manifestarse como cefalea, visión borrosa, somnolencia, edema papilar, hemorragias retinianas, epistaxis, sangrado gastrointestinal, púrpura e insuficiencia cardiaca. La proteína monoclonal , además se relaciona con la aparición de anticoagulante lúpico, deficiencia adquirida de la proteína S, resistencia adquirida a la proteína C e inhibición del activador tisular del plasminógeno.
Estudios en la Evaluación del Mieloma Múltiple
En la biometría hemática puedes encontrar una anemia normocítica normocrómica en los estadios iniciales. En estadios avanzados puede asociarse leucopenia con neutropenia y trombocitopenia. Los hematíes aparecen aglutinados en columnas o pilas de monedas (rouleaux) en el frotis de sangre periférica. En el aspirado de médula ósea destaca la infiltración por células plasmáticas. El núcleo es redondo u oval, con áreas de cromatina densa “en rueda de carro”. Las células plasmáticas en el mieloma múltiple expresan en la membrana CD38 y CD56, siendo la mayoría CD10, HLA-DR y CD20 negativas.
La proteína monoclonal sérica mayor de 30 g/L para IgG y mayor de 25 g/L para IgA ó mayor de 1 g/24 horas de cadenas ligeras en orina. En cuanto a las proteínas plasmáticas clonales en la médula ósea, éstas exceden el 10%. Las determinaciones de la concentración de la proteína C reactiva, LDH y β2-microglobulina pueden encontrarse por encima del rango de la normalidad, sobre todo en pacientes con mal pronóstico. No olvides realizar pruebas de función renal con electrolitos séricos ya que en la cuarta parte de los pacientes diagnosticados con mieloma múltiple aparece enfermedad renal con cifras de creatinina superiores a 2 mg/dl, secundaria a la hipercalcemia y al filtrado glomerular, y posterior acumulación tubular de cadenas ligeras.
En estos casos encontrarás elevada la urea y el ácido úrico. La fosfatasa alcalina puede encontrarse elevada en los pacientes con afectación ósea. En más de la mitad de los pacientes se identifica en el examen de orina la presencia de cadenas ligeras monoclonales (proteinuria de Bence-Jones). En el 15% solo se detecta una única banda monoclonal en la orina (mieloma Bence Jones). La radiografía convencional de la columna vertebral, cráneo, tórax, pelvis , húmero y fémur sigue siendo el estándar de oro para identificar lesiones líticas.
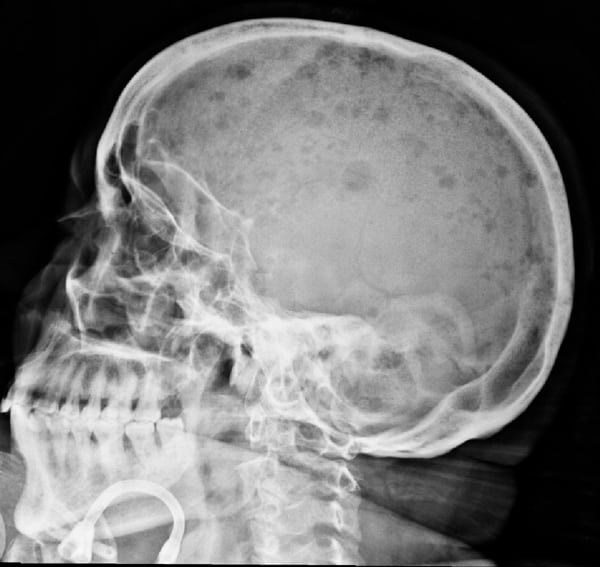
Se recomienda la resonancia magnética para evaluar los síntomas en pacientes con resultados normales en la radiografía convencional o en todos los pacientes con radiografías sugerentes de plasmocitoma solitario del hueso.
Pruebas Complementarias
La electroforesis de proteínas concentradas en la orina es muy útil porque diferencia las lesiones glomerulares de las lesiones tubulares. Las lesiones glomerulares, como las que se producen por depósitos glomerulares de amiloides o enfermedad de depósito de cadenas ligeras, dan por resultado la pérdida no selectiva de todas las proteínas séricas en la orina; la configuración de esta orina en la electroforesis se asemeja a la configuración sérica con un predominio de albúmina. La Citogenética mediante la hibridación in situ por fluorescencia (FISH) se utiliza específicamente para determinar las mutaciones 17p13, t(4;14), t(11;14)
Diagnóstico de Mieloma Múltiple
El diagnóstico requiere células plasmáticas de médula ósea clonales >10% o un plasmocitoma probado por biopsia más evidencia de uno o más eventos definidores de mieloma múltiple: CEREBRO (hipercalcemia, insuficiencia renal o lesiones líticas) características relacionadas con el trastorno de las células plasmáticas, plasmocitosis clonal de la médula ósea >60%, proporción sérica involucrada / sin compromiso de la cadena libre (FLC) >100 (siempre que el FLC involucrado sea >100mg/L) o >1 focal en la resonancia magnética
No toda gammapatía monoclonal es mieloma múltiple. Con este nombre se conocen una serie de procesos caracterizados por la expansión clonal de células linfoides, que en condiciones normales son las encargadas de las síntesis de inmunoglobulinas. Se incluyen algunos no malignos como la gammapatía monoclonal de significado incierto y malignos como la macroglobulinemia de Waldenström y la amiloidosis.
El plasmocitoma óseo solitario es un tumor localizado de células plasmáticas, histológicamente idénticas a aquellas que se ven en el mieloma múltiple, en ausencia de otras lesiones osteolíticas y de hallazgos diagnósticos del mieloma. Aproximadamente un 50% de los plasmocitomas desarrollan mieloma múltiple al cabo de 10 años. Si se presenta fuera del hueso se denomina plasmocitoma extramedular. El tratamiento de elección es la radioterapia, el tratamiento quirúrgico es para tratar el colapso vertebral y compresión medular o radicular.
El plasmocitoma extramedular es una de las formas más raras de presentación de la amplia patología tumoral de las células plasmáticas, ya que representa menos del 4% del total. Su localización más habitual es la cabeza y el cuello, en cerca del 90% de los casos.
Tratamiento del Mieloma Múltiple
El tratamiento inicial consiste en bortezomib, lenalidomida, dexametasona (VRD). En los pacientes de alto riesgo, carfilzomib, lenalidomida, dexametasona (KRD) es una alternativa. En los pacientes elegibles , el tratamiento inicial se administra durante aproximadamente 3-4 meses seguido de trasplante autólogo de células madre (ASCT). Los pacientes que no son candidatos para trasplante se tratan con radioterapia hasta la progresión, ó alternativamente, un régimen de tripletes como VRD durante aproximadamente 12-18 meses.
Después de ASCT, el mantenimiento de lenalidomida, se considera para pacientes de riesgo estándar, especialmente en aquellos que no están en respuesta parcial o mejor. El mantenimiento se realiza con un régimen basado en bortezomib. Los pacientes con recaída pueden ser tratados con una combinación de 2 fármacos o de 3 fármacos. Los pacientes con recaídas más agresivas requieren un régimen de tres fármacos.
Tratamientos en Investigación
El daratumumab, un anticuerpo monoclonal IgG que se dirige a CD38, induce la actividad anti mieloma directa e indirectamente y ha demostrado su eficacia como monoterapia en pacientes con mieloma múltiple. En un ensayo clínico fase III con 498 pacientes mostró una tasa de supervivencia en 12 meses libre de progresión del 60 % en el grupo de daratumumab frente al 26.9% en el grupo control. Entre los pacientes con mieloma múltiple recurrente o refractario, daratumumab en combinación con bortezomib y dexametasona resultó en una supervivencia libre de progresión significativamente más larga.
Pronóstico del Paciente con Mieloma Múltiple
El pronóstico de mieloma múltiple se determina por el número de células de mieloma y por las características del paciente. El factor pronóstico individual más importante corresponde a la concentración de β2-microglobulina; los valores altos pronostican una mortalidad temprana. A mayor edad y peor estado funcional (ECOG) , mayor tendencia a comorbilidades e infecciones y menor tolerancia a tratamientos intensivos.
Referencias Bibliográficas
Costello C. An update on the role of daratumumab in the treatment of multiple myeloma. Ther Adv Hematol. 2017;8(1):28-37.
Mulligan G, Lichter DI, Di bacco A, et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood. 2014;123(5):632-9.
Gerecke C, Fuhrmann S, Strifler S, Schmidt-hieber M, Einsele H, Knop S. The Diagnosis and Treatment of Multiple Myeloma. Dtsch Arztebl Int. 2016;113(27-28):470-6.
Mitchell JS, Li N, Weinhold N, et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat Commun. 2016;7:12050.
Kuhr K, Wirth D, Srivastava K, Lehmacher W, Hellmich M. First-line therapy for non-transplant eligible patients with multiple myeloma: direct and adjusted indirect comparison of treatment regimens on the existing market in Germany. Eur J Clin Pharmacol. 2016;72(3):257-65.




