Cáncer en Pediatría: Claves Diagnósticas de los más Frecuentes.
El cáncer constituye la segunda causa de muerte entre uno y los catorce años de edad, con picos de incidencia durante la primera infancia y la adolescencia. A continuación hacemos una breve revisión de los tipos de cáncer más frecuentes en pediatría.
Con menor frecuencia y siguiendo en orden descendente, se presentan los sarcomas de partes blandas, los tumores de células germinales y el retinoblastoma. Estas entidades, junto con los linfomas, las abordaremos en otras revisiones.
Leucemia
La leucemia es la neoplasia más frecuente en pediatría. El 97% son agudas, de las cuales el 77% son linfoblásticas y 20% mieloblásticas, el 3% restante son mieloides crónicas. En el siguiente cuadro resumimos los factores de riesgo para la leucemia.
| Genéticos | Ambientales |
|---|---|
| Síndrome de Down | Radiación Ionizante |
| Síndrome de Fanconi | Edad avanzada de la madre |
| Ataxia-Telangiectasia | Benceno |
| Síndrome de Schwachman | Hidrocarburos y pesticidas |
| Anemia de Blackfan-Diamond | Hermano gemelo con leucemia |
| Síndrome de Bloom | Alquilantes |
| Disqueratosis Congénita | Nitrosureas |
| Neurofibromatosis | |
| Síndrome de Turner | |
| Síndrome de Klinefelter | |
| Inmunodeficiencia Combinada Grave | |
| Síndrome de Li-Fraumeni |
Presentación Clínica
La leucemia de reciente inicio tiene clínica inespecífica, con cansancio, dolor óseo y/o articular, fiebre persistente, etc. Conforme avanza, aparecen las manifestaciones clínicas de falla medular, las cuales se dividen en 5 síndromes:
- Anémico: Palidez y síntomas hipóxicos como: fatiga, irritabilidad, astenia, adinamia y taquicardia.
- Neutropénico: Fiebre y/o procesos infecciosos persistentes o recurrentes.
- Purpúrico: Petequias, púrpura, equimosis, epistaxis, gingivorragia u otras manifestaciones de sangrado secundarias a trombocitopenia.
- Infiltrativo: Dolor óseo, adenomegalias, hepatomegalia, esplenomegalia, infiltración a piel, parótidas, encías o testículos, con formación de tumores sólidos, leucocitosis y masa mediastinal.
- Metabólico: Alteraciones bioquímicas por la carga tumoral total y consecuencia de la proliferación y destrucción excesiva de células leucémicas. Se manifiestan como hiperuricemia, hiperkalemia, hipocalcemia, hiperfosfatemia, elevación de la creatinina (síndrome de lisis tumoral) y elevación de la deshidrogenasa láctica.
Estudios que serán de utilidad
Ahora que ya tienes por clínica enfocado tu diagnóstico, te puedes apoyar de estudios de laboratorio.
- Biometría hemática completa
- Frotis de sangre periférica
- Química sanguínea
- Electrolitos séricos (Na, K, Ca, Mg)
- Pruebas de función hepática
- Pruebas de coagulación
- Aspirado de médula ósea
- Estudio de líquido cefalorraquídeo
- Inmunofenotipo en médula ósea
- Cariotipo en médula ósea.
La biometría hemática puede ser normal en la etapa de leucemia reciente. Durante la evolución pueden aparecer citopenias aisladas (anemia, neutropenia o trombocitopenia) o pancitopenia. Se observa la aparición de blastos en el frotis de sangre periférica en etapa tardía.
En los estudios de imagen solicita rayos X, en la que puede aparecer una masa en mediastino anterior en 30-50% de los pacientes. La radiografía de huesos largos puede ser normal o presentar lesiones mínimas inespecíficas en la etapa inicial y sólo en etapas avanzadas se aprecian lesiones óseas francas. El ultrasonido (US) testicular se realiza en aquellos niños con crecimiento de testículos por la posibilidad de infiltración testicular.
Tratamiento y Pronóstico
El tratamiento depende del tipo de leucemia y riesgo estimado de cada paciente. La mayoría de los protocolos tienen varias fases. La primera es la de inducción de la remisión, durante la que se intenta eliminar las células de la médula ósea. Se utiliza vincristina, L-asparaginasa y un corticoide; en ocasiones también quimioterapia intratecal. Se considera que el paciente entra en remisión con menos de 5% de blastos en médula ósea. La segunda fase va enfocada al sistema nervioso central, para después administrar quimioterapia por tiempo prolongado hasta la fase de mantenimiento.
Se considera de buen pronóstico la hiperdiploidía, la translocación TEL/AML1, así como la buena respuesta a la quimioterapia. La recidiva se considera el principal factor de mal pronóstico. Otros factores considerados de mal pronóstico son:
- Edad menor a un año o mayor de 10.
- Recuento leucocitario > 100,000/µL.
- Hipodiploidía, cromosoma Filadelfia o t(4;11).
- Respuesta lenta al tratamiento inicial.
Tumores Cerebrales
Los tumores cerebrales son el segundo tipo de cáncer más frecuente en la infancia, con una alta morbi- y mortalidad y mayor incidencia durante la lactancia y en niños pequeños. Los más comunes son los astrocitomas y el meduloblastoma. La siguiente tabla relaciona los síndromes reconocidos como factores de riesgo y los tumores asociados.
| Síndrome de Cowden | Gangliocitoma displásico del cerebelo |
|---|---|
| Síndrome de Li-Fraumeni | Astrocitoma, meduloblastoma |
| Neurofibromatosis tipo I | Glioma óptico, astrocitoma, neurofibroma. |
| Neurofibromatosis tipo II | Schwanoma vestibular, meningioma, ependimoma, astrocitoma, hamartoma. |
| Esclerosis tuberosa | Astrocitoma de células gigantes subependimario. |
| Síndrome de Turcot | Meduloblastoma |
| Sx. de von Hippel-Lindau | Hemangioblastoma |
Presentación Clínica
La clínica de los tumores cerebrales depende de la localización, el tipo de tumor y la edad del paciente. La presentación puede ser precedida por cambios sutiles de la personalidad, alteración del lenguaje y capacidades mentales. Sospecha de un tumor cerebral ante un paciente pediátrico pequeño que refiere cefalea, la cual no cede a la analgesia, de predominio matutino y asociada a vómitos, pérdida de peso u otros síntomas constitucionales.
Los tumores que obstruyen el drenaje del líquido cefalorraquídeo (LCR) pueden ocasionar clínica de hipertensión intracraneal, así como abombamiento de las fontanelas en lactantes. Los tumores de la línea media o infratentoriales ocasionan la clásica triada de cefalea, vómitos y edema papilar; asocian en algunos casos alteraciones de la marcha y el equilibrio.
En los casos con compromiso del tallo cerebral, la clínica es secundaria a la parálisis de pares craneales, así como déficit de la neurona motora superior. Por último, los tumores supratentoriales ocasionan déficits focales, motores y convulsiones.
Diagnóstico
Ante la sospecha clínica debes dirigir tu anamnesis y realizar exploración neurológica, así como interconsultar a oftalmología. La resonancia magnética es el estudio de elección. El diagnóstico definitivo se realiza mediante el estudio histopatológico de la pieza quirúrgica, mientras que en los casos no resecables se puede hacer una biopsia por estereotaxia. En tumores con extensión leptomeníngea se debe realizar una punción lumbar, como p.e. en el meduloblastoma, ependimoma y tumores germinales.
Al momento de la exploración física debes enfocarte en los hallazgos de la exploración neurológica, examinando pares craneales, marcha, sensibilidad, función neuromuscular, cerebelo-vestibular y reflejos osteotendinosos. Siempre estima el Glasgow (modificado; siguiente tabla) y explora fondo de ojo.
| Actividad | Respuesta |
|---|---|
| Verbal Balbucea, llora, ríe Llanto continuo Llanto exagerado Gruñidos Ausencia | 5 4 3 2 1 |
| Motora Movimientos espontáneos Localiza dolor Retira al dolor Flexión anormal Extensión anormal Ausencia | 5 4 3 2 1 |
| Apertura de ojos Espontánea Al hablarle Al dolor Ausencia | 4 3 2 1 |
| Escala de Glasgow Modificada (Lactantes) | |
Astrocitomas
Los astrocitomas representan el 40% de todas las neoplasias del Sistema Nervioso Central (SNC) en pediatría. Un 15 a 25% son de bajo grado y 10 a 15% de alto grado. Los factores de riesgo reconocidos para desarrollar astrocitoma son la neurofibromatosis, esclerosis tuberosa, síndrome de Von-Hippel Lindau, síndrome Li-Fraumeni y de Turcot, éste último también denominado síndrome de tumor cerebral y poliposis. (ver tabla anterior).
Los astrocitomas de bajo grado tienen una presentación subclínica, siendo el astrocitoma pilocítico juvenil el más frecuente de este grupo y generalmente localizado en cerebelo. Se clasifica como de grado I, según la OMS, y en la RM se observa como un nódulo con captación de contraste. En el estudio histológico se observan fibras de Rosenthal. El astrocitoma fibrilar de bajo grado es el segundo más frecuente y se clasifica de grado II, según la OMS. La RM no capta contraste y es de peor pronóstico que el pilocítico.
El tratamiento de elección de los astrocitomas de bajo grado es la resección quirúrgica completa, con una mortalidad de menos del 20%. En caso de irresecabilidad o cuando ésta es parcial está indicada la radioterapia. Se hace uso de quimioterapia, mediante carboplatino y vincristina, para aumentar las tasas de curación y reducir la dosis total de radioterapia o diferirla, en especial en niños menores de 3 años.
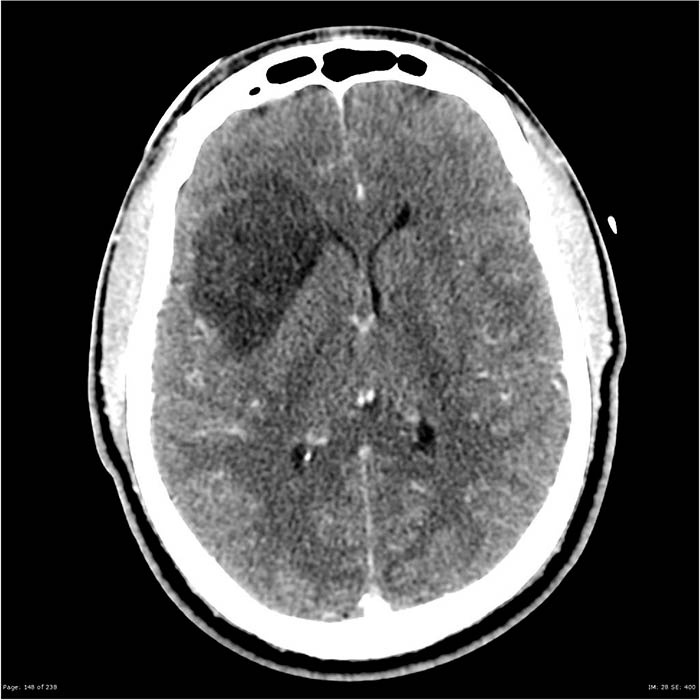
Astrocitomas de Alto Grado
Son poco frecuentes en pediatría, siendo el astrocitoma anaplásico el más frecuente; se considera grado III de la OMS. El glioblastoma multiforme es clasificado grado IV de la OMS. El tratamiento de este grupo de tumores es la resección quirúrgica radical con determinación de p53. Posterior a la cirugía se recomienda la radioterapia y quimioterapia con lomustina, prednisona y vincristina.
Meduloblastoma
El meduloblastoma forma parte de los tumores neuroectodérmicos primitivos, siendo el más frecuente de este grupo. Predomina en varones entre los 5 y 7 años de edad y se localiza por lo general en la vermis cerebelosa. Ocasiona clínica de hipertensión intracraneal y disfunción cerebelosa. Hasta un tercio de los casos presentan diseminación leptomeníngea. Por tanto, está indicado realizar estudios de imagen contrastados, tanto cerebral y medular, así como punción lumbar.
Se observa como una masa sólida y homogénea que realza con contraste. Los niños menores de 4 años, aquellos con enfermedad diseminada al momento del diagnóstico y tumor residual posterior a la cirugía tienen el peor pronóstico. Por el contrario, aquellos con aumento de la expresión del receptor de la neurotrofina (TRkb) y una baja expresión del oncogen MYC tienen mejor pronóstico.
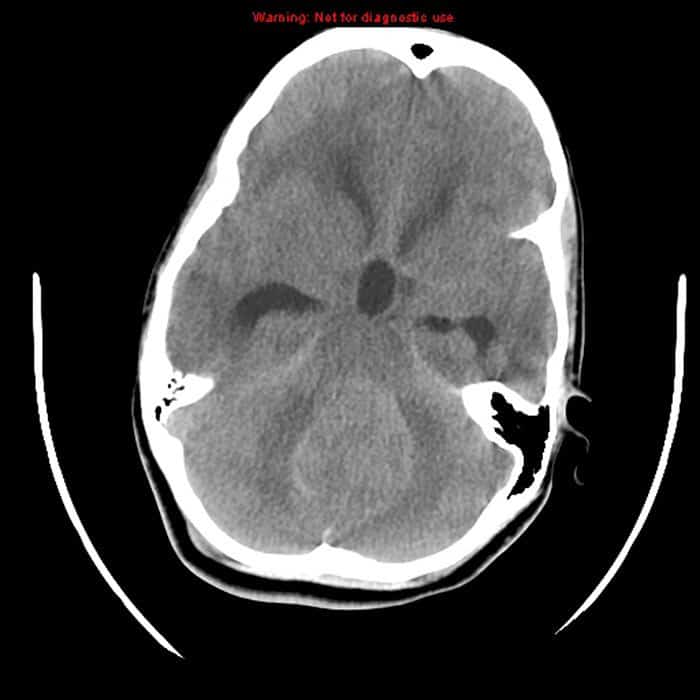
El tratamiento consiste en la cirugía, con el objetivo de estadificar el tumor y extirpar la mayor masa tumoral posible, procurando evitar lesiones neurológicas importantes. Posterior a la cirugía se indica radioterapia, añadiendo quimioterapia en pacientes de alto riesgo, como p.e. menores de 3 años, citología positiva en LCR y restos tumorales > 1.5 cm. Los quimioterapéuticos más eficaces son las nitrosoureas, vincristina y derivados del platino.
Masas Abdominales
Considera siempre cualquier masa abdominal en un niño como maligna hasta no demostrar lo contrario. Es importante recabar factores de riesgo durante la anamnesis, tales como exposición a radiación ionizante, quimioterapia, síndrome de Beckwith-Wiedemann, de Denys-Drash o de Sotos.
Ante el hallazgo durante la exploración física de tu paciente de una masa abdominal palpable, y/o presencia de dolor abdominal persistente, distensión, alteración de los hábitos evacuatorios, vómitos persistentes o hematuria, solicita una radiografía simple de abdomen o ecografía. En caso de hallazgos patológicos deberás referir a tu paciente a segundo nivel.
Neuroblastoma
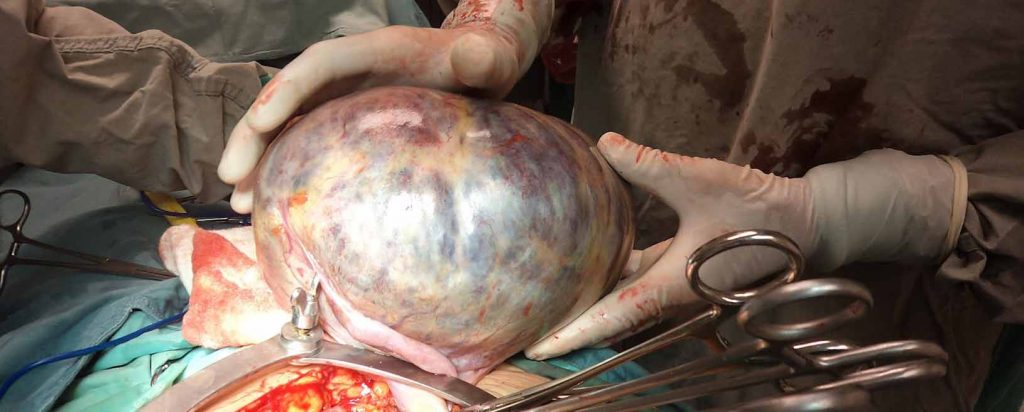
El neuroblastoma es un tumor maligno embrionario del sistema nervioso periférico con predominio en varones. Se trata del tumor sólido extracraneal más frecuente en pediatría, siendo la edad media al momento del diagnóstico de dos años de edad y el 90% diagnosticado antes de los cinco años. La localización más frecuente es la abdominal (70%), con 45% de estos casos localizados en la glándula suprarrenal.
Otras posibles localizaciones son la torácica y en mediastino posterior (20%), así como nasofaringe (estesioneuroblastoma; 4%). El neuroblastoma se asocia al síndrome alcohólico fetal, hidantoínas, neurofibromatosis tipo I, nesidioblastosis y enfermedad de Hirschsprung. De igual forma, existen formas familiares con herencia autosómica dominante.
Presentación Clínica
La clínica depende de la localización del neuroblastoma, de las metástasis a distancia, así como de la presencia de síndromes complejos con liberación de sustancias producidas por el tumor. Cuando se localiza en abdomen, se palpa una masa firme y nodular (70% de los casos), y hepatomegalia, dependiendo de la presencia o no de metástasis. En los tumores paraespinales la clínica es de compresión medular, con dolor radicular, disfunción genitourinaria y/o gastrointestinal, paraplejia, etc.
Cuando se localiza en tórax, el neuroblastoma suele ser un hallazgo incidental en la radiografía de tórax. En ocasiones puede ocasionar obstrucción mecánica o síndrome de vena cava superior. En cabeza y cuello puede generar síndrome de Horner, mientras que en nasofaringe se manifiesta por epistaxis. La metástasis ocurre vía linfática o hemática, principalmente a hígado, médula ósea y hueso. Los síndromes que derivan de dicha metástasis son:
- Síndrome de Hutchinson ante metástasis en médula ósea y hueso, con dolor óseo.
- Hipertensión intracraneal ante la infiltración de la duramadre.
- Síndrome de Smith en la afectación de la piel.
- Síndrome de Pepper ante la metástasis a hígado y más frecuente en lactante y recién nacidos.
Síndromes Complejos
El síndrome de mioclono-opsoclono o de Kinsbourne se presenta con ataxia, mioclono, opsoclono y demencia progresiva. Se presenta en casos con neuroblastoma abdominal o torácico y no afecta al cerebro. El síndrome de Kerner-Morrison es secundario a la producción de péptido intestinal vasoactivo (VIP) y cursa con hipopotasemia y deshidratación. Por otro lado, en casos raros se puede presentar un cuadro de hipertensión arterial y diaforesis por liberación de catecolaminas. Por último, en algunos pacientes con neuroblastoma se puede observar un hematoma lineal palpebral.
Estudios que serán de utilidad
La ecografía abdominal es de utilidad y la primera prueba a realizar si la localización del neuroblastoma es abdominal. Brinda información sobre el tamaño, tipo de masa y su localización. La TAC y RM abdominal y torácica ayudan a delimitar el tumor y descartar metástasis pulmonar. Se puede observar un tumor de densidad mixta con elementos sólidos y zonas de hemorragia o necrosis, así como calcificaciones en el 80% de los casos.
Puedes indicar determinación de catecolaminas en orina de 24 hrs. en tumores suprarrenales, así como gammagrafía con MIBG para el diagnóstico de extensión. La tomografía por emisión de positrones está indicada únicamente en estadios avanzados y MIBG negativa, síndrome de Kinsbourne sin tumor primario, o para descartar la presencia de enfermedad ganglionar abdominal persistente estadio III posterior al tratamiento.
Diagnóstico Anatomopatológico
El diagnóstico definitivo se establece mediante la toma de biopsia y estudio histológico de la muestra. El neuroblastoma se deriva de células de la cresta neural con grado variable de diferenciación. El pronóstico, acorde al diagnóstico histopatológico, depende de la cantidad de estroma, grado de diferenciación y mitosis. El parénquima tumoral tiende a la hemorragia, con zonas de necrosis y calcificación. Se debe determinar la amplificación del oncogén N-MYC.
Para los tumores que parecen estar localizados y resecables sin una morbilidad sustancial, el procedimiento de diagnóstico inicial puede incluir una resección completa o casi completa del tumor primario y un muestreo de los ganglios linfáticos ipsilaterales y contralaterales no adherentes.
Estadificación del Neuroblastoma
- Estadio I: Localizado.
- Estadio II. Situado más allá de la estructura de origen, sin sobrepasar la línea media y sin afectación ganglionar ipsilateral (IIA) o con ella (IIB).
- Estadio III: Más allá de la línea media con o sin afectación ganglionar.
- Estadio IV: Metástasis a distancia.
- Estadio IVs: Niños < 1 año de edad con tumor primario en estadio I o II con metástasis en hígado, piel o médula ósea.
Factores Pronósticos
La edad menor a un año al momento del diagnóstico, así como estadios I, II y IVs, así como la ausencia de oncogén N-Myc son considerados factores de buen pronóstico. La mortalidad global es del 26%, mientras que en pacientes de alto riesgo es del 70%. Se debe dar seguimiento estrecho a largo plazo a todos los pacientes en remisión de neuroblastoma, debido a los efectos adversos del tratamiento.
Tratamiento
El tratamiento en pacientes con bajo riesgo, es decir, estadios I, II y IVs es la resección quirúrgica. Los pacientes con riesgo intermedio son manejados mediante cirugía y quimioterapia, asociando en ocasiones radioterapia. Los pacientes de alto riesgo, es decir, estadio IV mayores de un año de edad, reciben quimioterapia de inducción. Posteriormente se les administra quimioterapia en dosis alta y se realiza trasplante de progenitores hematopoyéticos.
Ante la recaída local en los estadios I, II y IVs se vuelve a intervenir al paciente quirúrgicamente, mientras que en los pacientes en estadio III con primera recaída se indica quimioterapia y radioterapia ante recidiva local. Se vuelve a intentar la resección completa. En estadio IV se administra quimioterapia.
Tumor de Wilms
Se trata del tumor renal más frecuente (80%) y el segundo abdominal maligno con mayor prevalencia durante la infancia. La edad de presentación es de uno a cinco años de edad, con pico entre los dos y tres años. Se asocia en algunos casos al gen del tumor de Wilms (WT1) en el cromosoma 11p, el cual codifica un factor de transcripción del riñón. El tumor de Wilms se asocia a aniridia, hemihipertrofia y malformaciones genitourinarias. Se asocia de igual manera a múltiples síndromes, como el síndrome de Denys-Drash, síndrome WAGR (Wilms, Aniridia, malformaciones Genitourinarias y Retraso mental), Sx. de Beckwith-Wiedemann, Sx. de Sotos y otros síndromes de hipercrecimiento.
La variante histológica favorable del tumor de Wilms se caracteriza por predominio de células epiteliales y elementos del estroma. Por el contrario, la desfavorable es anaplásica, con núcleos hipercromáticos y aumento en el número de mitosis. Además, en el 85% de los casos presenta mutación del gen de supresión tumoral p53. Por último, el sarcoma de células claras es de muy mal pronóstico, con tendencia a metastatizar a hueso.
Presentación Clínica
El tumor de Wilms se presenta como una masa abdominal asintomática en el 75% de los casos, la cual se sitúa en un flanco, es redondeada, de consistencia elástica y no suele superar la línea media. El 60% de los pacientes se presentan con hipertensión arterial por compresión de la arteria renal y producción de renina. Se observa hematuria micro- y macroscópica, así como policitemia por producción de eritropoyetina. El lugar más frecuente de metástasis son los ganglios regionales y los pulmones, observándose en el 10 a 15% de los pacientes al momento del diagnóstico.
Estudios que serán de utilidad
La ecografía abdominal permite la localización del tumor, mientras que la TAC y RM abdominal son de utilidad para corroborar el origen intrarrenal del tumor, su extensión, afectación de la vena cava e integridad del riñón contralateral. La radiografía y TAC de tórax permiten identificar las metástasis pulmonares. No está indicada la toma de biopsia dado que la ruptura de la cápsula renal cambia el estadio tumoral. La gammagrafía ósea está indicada en el sarcoma de células claras, dado que tiende a metastatizar a hueso.
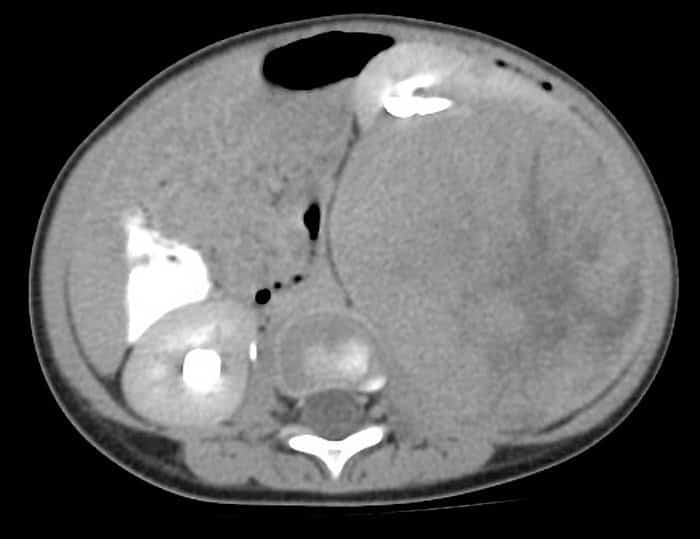
Estadificación del Tumor de Wilms
- Estadio I: Limitado al riñón, con cápsula íntegra y extirpación completa.
- Estadio II: Crecimiento más allá del riñón, pero con cápsula íntegra y extirpación completa.
- Estadio III: Restos tumorales postquirúrgicos, sin afectación hematógena.
- Estadio IV: Metástasis hematógena.
- Estadio V: Afectación bilateral.
Tratamiento
El tratamiento depende del estadio y el tipo histológico. Se realiza resección quirúrgica, con previa valoración de la permeabilidad de la vena cava inferior. Se administra quimioterapia prequirúrgica si no está permeable. En el estadio I, de riesgo moderado o alto, se administra quimioterapia postoperatoria (generalmente vincristina y actinomicina D).
En los estadios II y III de riesgo bajo y moderado se utiliza un protocolo similar; en los pacientes de alto riesgo se añaden usualmente doxorrubicina y ciclofosfamida, entre otros quimioterapéuticos, junto con radioterapia. En el estadio IV se administran también varios agentes quimioterapéuticos, así como radioterapia (tanto abdominal como torácica).
En los tumores bilaterales se administra quimioterapia para reducir el tamaño del tumor y lograr la máxima resección posible, con nefrectomía completa unilateral y parcial del riñón menos afectado. Se consideran factores de buen pronóstico un tipo histológico favorable, los estadios I y II, edad menor de dos años y una masa tumoral pequeña. La mortalidad global de los tumores renales es del 10% a cinco años. La anaplasia en la histología confiere un muy mal pronóstico.
Nefroma Mesoblástico
El tumor de Bolande o mesoblástico es el tumor congénito renal más frecuente, suele ser benigno, predomina en varones y deriva de tejidos fetales, con diagnóstico en el periodo neonatal. Se presenta como una gran masa renal productora de renina. El tratamiento es quirúrgico.
Tumores óseos
El tumor óseo más frecuente de la infancia es el osteosarcoma. En recién nacidos la neoplasia ósea más frecuente es la metástasis de un neuroblastoma. En menores de 10 años es más frecuente el sarcoma de Ewing.
Osteosarcoma
El osteosarcoma es el tumor óseo primario más frecuente en niños y adolescentes. Suele presentarse en las etapas de máximo crecimiento, por lo general durante la segunda década de la vida. Se asocia a retinoblastoma, síndrome de Li-Fraumeni y de Rothmund-Thomson. La localización más frecuente es la metáfisis femoral distal y tibia proximal. El osteosarcoma parostal es de bajo grado, bien diferenciado, no invade la médula ósea y no suele metastatizar. El osteosarcoma periostal se origina de la superficie del hueso y tiende a la metástasis.
Presentación Clínica
El paciente refiere dolor y tumefacción. Durante la anamnesis enfatiza sobre los síntomas y sus características, como la duración, intensidad y tiempo de presentación, dolor nocturno o fracturas previas. El dato pivote que se reporta en el 100% de los pacientes es dolor en el sitio primario del tumor (localizado). A la exploración física busca:
- Aumento de volumen: masa visible, asociada a datos de inflamación: calor y rubor.
- Piel adelgazada y brillosa (por distensión).
- Red venosa colateral.
- Localización y limitación en la movilidad.
- Relación del edema con involucramiento del tejido blando y óseo.
- Presencia de linfadenopatía regional/local.
Estudios que serán de utilidad
Realiza una evaluación radiográfica, que es la herramienta fundamental para el diagnóstico. Se observa una lesión agresiva en metáfisis distal de huesos largos, con destrucción del patrón trabecular, así como áreas radiopacas con formación de hueso nuevo y reacción perióstica agresiva, conocida como “triángulo de Codman” ó la famosa imagen en “sol naciente” asociada a los tejidos blandos aledaños.
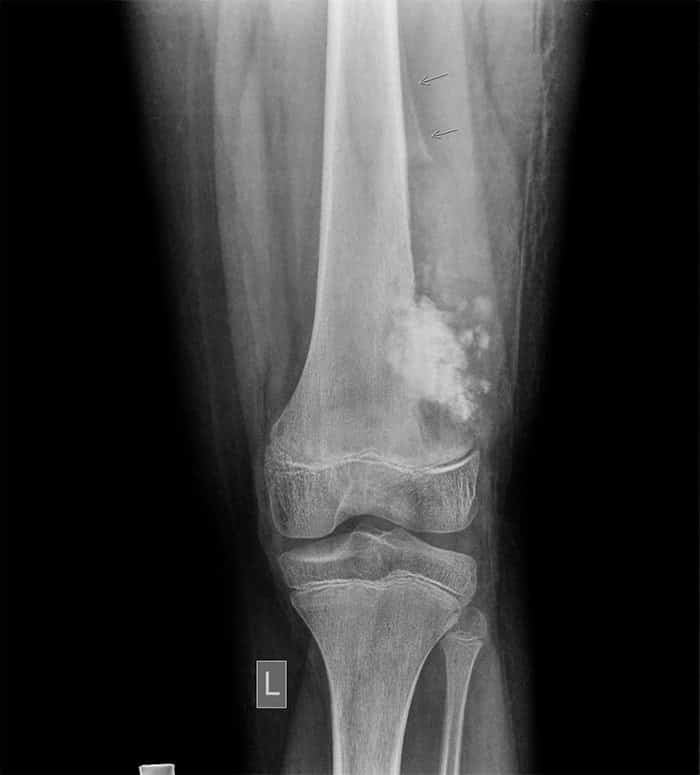
Se debe realizar una resonancia del hueso afectado con toma de biopsia, así como estudio de extensión para descartar metástasis torácicas (mediante TAC) y óseas (gammagrafía).
Tratamiento y Pronóstico
Se realiza la resección quirúrgica con márgenes amplios, junto con quimioterapia pre- y postoperatoria. La supervivencia a 5 años es del 75%. Las metástasis pulmonares deben resecarse. Los factores de mal pronóstico son el hallazgo de metástasis al momento del diagnóstico, mala respuesta a la quimioterapia preoperatoria, expresión del gen MDR, así como valores elevados de fosfatasa alcalina y LDH.
Sarcoma de Ewing
El sarcoma de Ewing es un tumor indiferenciado de células pequeñas provenientes de la cresta neural y asociado en la mayoría de los casos a la t(11;22). Los marcadores tumorales de este tumor son la proteína S-100 o la enolasa neuroespecífica. Al igual que el osteosarcoma, se presenta durante la segunda década de vida y suele localizarse en la diáfisis de huesos largos.
El paciente refiere dolor y tumefacción, asociados a síntomas constitucionales, tales como con fiebre, malestar y pérdida de peso. En la radiografía se observa una lesión lítica primaria con reacción perióstica o en “piel de cebolla”. Los estudios de extensión incluyen TAC de tórax, gammagrafía ósea y biopsia de médula ósea. El tratamiento incluye la resección quirúrgica aunada a quimioterapia pre- y postoperatoria. En algunos casos se asocia radioterapia.
| Característica | Osteosarcoma | Sarcoma de Ewing |
|---|---|---|
| Edad | Segunda década | Segunda década |
| Células | Fusiforme, productora de osteoide. | Pequeña, redonda e indiferenciada. Presenta marcadores tumorales. |
| Asociaciones | Retinoblastoma, Li- Fraumeni, radioterapia, Rothmund-Thomson | No conocidas. |
| Localización | Metáfisis de huesos largos (fémur o tibia). | Diáfisis de huesos largos, huesos planos. |
| Clínica | Dolor más tumefacción. | Dolor más tumefacción y síntomas constitucionales. |
| Signos radiológicos | Signo de sol naciente y triángulo de Codman. | Patrón en piel de cebolla. |
Referencias Bibliográficas
Kliegman, R., Behrman, R.E, Stanton, Game, S. & Schor. (2017). Nelson tratado de pediatría. España: Elsevier.
WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, revised 4th edition, Swerdlow SH, Campo E, Harris NL, et al. (Eds), International Agency for Research on Cancer (IARC), Lyon 2017.
Clarke RT, Van den Bruel A, Bankhead C, et al. Clinical presentation of childhood leukaemia: a systematic review and meta-analysis. Arch Dis Child 2016; 101:894.
Margolin JF, Steuber CP, Poplack DG. Acute Lymphoblastic Leukemia. In: Principles and Practice of Pediatric Oncology, 4th ed, Pizzo PA, Poplack DG (Eds), Lippincott-Raven, Philadelphia 2001. p.489.
Bleyer WA. Central nervous system leukemia. Pediatr Clin North Am 1988; 35:789.
Ingram LC, Fairclough DL, Furman WL, et al. Cranial nerve palsy in childhood acute lymphoblastic leukemia and non-Hodgkin’s lymphoma. Cancer 1991; 67:2262.
Paryani SB, Donaldson SS, Amylon MD, Link MP. Cranial nerve involvement in children with leukemia and lymphoma. J Clin Oncol 1983; 1:542.
Liu HC, Yeh TC, Hou JY, et al. Triple intrathecal therapy alone with omission of cranial radiation in children with acute lymphoblastic leukemia. J Clin Oncol 2014; 32:1825.
Vora A, Andreano A, Pui CH, et al. Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol 2016; 34:919.
Paolucci G, Vecchi V, Favre C, et al. Treatment of childhood acute lymphoblastic leukemia. Long-term results of the AIEOP-ALL 87 study. Haematologica 2001; 86:478.
Wilson PE, Oleszek JL, Clayton GH. Pediatric spinal cord tumors and masses. J Spinal Cord Med 2007; 30 Suppl 1:S15.
Wilne S, Koller K, Collier J, et al. The diagnosis of brain tumours in children: a guideline to assist healthcare professionals in the assessment of children who may have a brain tumour. Arch Dis Child 2010; 95:534.
Matthews PM, Wylezinska M, Cadoux-Hudson T. Novel approaches to imaging brain tumors. Hematol Oncol Clin North Am 2001; 15:609.
Houten JK, Babu RP, Miller DC. Thoracic paraganglioma presenting with spinal cord compression and metastases. J Spinal Disord Tech 2002; 15:319.
Houten JK, Weiner HL. Pediatric intramedullary spinal cord tumors: special considerations. J Neurooncol 2000; 47:225.
Merchant TE, Kiehna EN, Thompson SJ, et al. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg 2000; 32:30.
Morris EB, Gajjar A, Okuma JO, et al. Survival and late mortality in long-term survivors of pediatric CNS tumors. J Clin Oncol 2007; 25:1532.
Armstrong GT, Liu Q, Yasui Y, et al. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst 2009; 101:946.
Blaney SM, Hass-Kogan D, Poussaint TY, et al. Gliomas, ependymomas, and other nonembryonal tumors of the central nervous system. In: Principles and practice of pediatric oncology, 6th ed, Pizzo P, Poplack D (Eds), Lippincott Williams & Wilkins, Philadelphia 2011. p.717.
Hall WA, Truwit CL. 1.5 T: spectroscopy-supported brain biopsy. Neurosurg Clin N Am 2005; 16:165.
Ahn ES, Goumnerova L. Endoscopic biopsy of brain tumors in children: diagnostic success and utility in guiding treatment strategies. J Neurosurg Pediatr 2010; 5:255.
Brodeur GM, Hogarty MD, Mosse YP, Maris JM. Neuroblastoma. In: Principles and Practice of Pediatric Oncology, Pizzo PA, Poplack DG (Eds), Lippincott Williams & Wilkins, Philadelphia 2011. p.886.
Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med 2015; 66:49.
Neuroblastic tumours of adrenal gland and sympathetic nervous system. In: Pathology and Genetics of Tumours of the Nervous System, World Health Organization, IARC, Lyon 2000. p.153.
DuBois SG, Kalika Y, Lukens JN, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol 1999; 21:181.
Mahoney NR, Liu GT, Menacker SJ, et al. Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 2006; 142:651.
Wilfong AA, Parke JT, McCrary JA 3rd. Opsoclonus-myoclonus with Beckwith-Wiedemann syndrome and hepatoblastoma. Pediatr Neurol 1992; 8:77.
Verma A, Brozman B. Opsoclonus-myoclonus syndrome following Epstein-Barr virus infection. Neurology 2002; 58:1131.
Picco P, Garaventa A, Claudiani F, et al. Primary hypothyroidism as a consequence of 131-I-metaiodobenzylguanidine treatment for children with neuroblastoma. Cancer 1995; 76:1662.
Garaventa A, Guerra P, Arrighini A, et al. Treatment of advanced neuroblastoma with I-131 meta-iodobenzylguanidine. Cancer 1991; 67:922.
Fernandez C, Geller JI, Ehrlich PF, et al. Renal tumors. In: Principles and Practice of Pediatric Oncology, 6th ed, Pizzo P, Poplack D (Eds), Lippincott Williams & Wilkins, St. Louis 2011. p.861.
Zuppan CW, Beckwith JB, Luckey DW. Anaplasia in unilateral Wilms’ tumor: a report from the National Wilms’ Tumor Study Pathology Center. Hum Pathol 1988; 19:1199.
Ganguly A, Gribble J, Tune B, et al. Renin-secreting Wilms’ tumor with severe hypertension. Report of a case and brief review of renin-secreting tumors. Ann Intern Med 1973; 79:835.
Ritchey ML, Azizkhan RG, Beckwith JB, et al. Neonatal Wilms tumor. J Pediatr Surg 1995; 30:856.
Tabori U, Sung L, Hukin J, et al. Medulloblastoma in the second decade of life: a specific group with respect to toxicity and management: a Canadian Pediatric Brain Tumor Consortium Study. Cancer 2005; 103:1874.
Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 2010; 28:1345.
Smoll NR, Drummond KJ. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J Clin Neurosci 2012; 19:1541.
Waszak SM, Northcott PA, Buchhalter I, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 2018; 19:785.
Huvos A. Bone Tumors: Diagnosis, Treatment, Prognosis, 2nd, WB Saunders, Philadelphia 1991.
Sissons HA. The WHO classification of bone tumors. Recent Results Cancer Res 1976; :104.
McKenna R, Schwinn C, Soong K, et al. Sarcomas of the osteogenic series (osteosarcoma, chondrosarcoma, parosteal osteogenic sarcoma, and sarcomata arising in abnormal bone): an analysis of 552 cases. J Bone Joint Surg Am 1966; 48:1.
Gorlick R, Bielack S, Teot L, et al. Osteosarcoma: Biology, diagnosis, treatment, and remaining challenges. In: Principles and Practice of Pediatric Oncology, 6th ed, Pizzo PA, Poplack DG (Eds), Lippincott, Williams and Wilkins, Philadelphia PA 2011. p.1015.
Buckley JD, Pendergrass TW, Buckley CM, et al. Epidemiology of osteosarcoma and Ewing’s
Ginsberg JP, Woo SY, Hicks MJ, Horowitz ME. Ewing’s sarcoma family of tumors: Ewing’s sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. In: Priniciples and Practice of Pediatric Oncology, 4th, Pizzo PA, Poplack DG (Eds), Lippincott, Williams and Wilkins, Philadelphia 2002.
Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol 2000; 18:3108.
Raney RB, Asmar L, Newton WA Jr, et al. Ewing’s sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol 1997; 15:574.