Enfermedad de Hirschsprung o Megacolon Agangliónico Congénito.

La enfermedad de Hirschsprung es un trastorno motor del intestino, causado por una migración incompleta de las células de la cresta neural (precursoras de las células del ganglio entérico) durante el desarrollo intestinal fetal. El segmento agangliónico resultante del colon no se relaja, causando una obstrucción funcional. Revisamos a continuación lo más relevante que debes conocer de su diagnóstico y tratamiento.
En aproximadamente el 80% de los pacientes, el trastorno afecta al recto y parte del colon sigmoide (conocido como enfermedad de segmento corto). En el 15 a 20% de los pacientes, la aganglionosis se extiende de manera proximal a lo largo del colon sigmoide (conocida como enfermedad de segmento largo). Por último, en aproximadamente el 5%, todo el colon está afectado (lo que se conoce como aganglionosis colónica total), y en casos raros el intestino delgado también puede estar involucrado.
Epidemiología
La enfermedad de Hirschsprung ocurre en aproximadamente 1 de cada 5000 nacidos vivos con una proporción general de hombres:mujeres de 3:1 a 4:1; cuando todo el colon está involucrado, la proporción de género se acerca más a 1:1. Existe un agrupamiento familiar para la enfermedad de Hirschsprung no sindrómica, con un riesgo de recurrencia global de aproximadamente el 3% en los hermanos para la enfermedad de segmento corto, o hasta el 17% si el probando tiene la enfermedad del segmento largo. Este riesgo de recurrencia es mayor si el probando es femenino, y también es mayor si se ven afectados varios miembros de la familia.
Etiología
La teoría más aceptada de la etiología de la enfermedad de Hirschsprung es que existe un defecto en la migración craneocaudal de neuroblastos originados en la cresta neural, un proceso que comienza a las cuatro semanas de gestación y termina en la semana 7 con la llegada de células derivadas de la cresta en el extremo distal del colon. El hecho de que las células no alcancen el colon distal deja ese segmento agangliónico y, por tanto, no funcional, lo que resulta en la enfermedad de Hirschsprung. Los defectos en la diferenciación de los neuroblastos en células ganglionares y su destrucción en el intestino también pueden contribuir al trastorno.
Genética
Se han identificado al menos 12 mutaciones genéticas en pacientes con enfermedad de Hirschsprung. El gen predominante afectado es el protooncogén RET, con mutaciones que causan pérdida de función. Las mutaciones de secuencia codificante en RET se identifican en aproximadamente la mitad de todos los casos familiares y en aproximadamente un tercio de los casos esporádicos. En un estudio, se encontraron variaciones de RET en el 82% de los pacientes con aganglionosis colónica total, en comparación con el 33% de aquellos con enfermedad de segmento corto. Se han descrito más de 20 mutaciones diferentes en el protooncogén RET. Además, ciertos polimorfismos RET están asociados con un fenotipo particular de enfermedad de Hirschsprung (enfermedad de segmento corto o largo).
La mayoría de los casos de Hirschsprung están vinculados a RET, incluso sin una mutación de secuencia codificadora identificada, lo que sugiere que las variantes no codificantes de este gen desempeñan un papel importante en la enfermedad. Una variante común en una secuencia similar a un potenciador en el intrón 1 del gen RET se ha asociado fuertemente con la enfermedad de Hirschsprung. También se ha encontrado una asociación fuerte entre un cierto haplotipo que comienza en la región promotora de RET y se extiende hasta el exón 2, y esto puede explicar muchos casos esporádicos en los que no se identifican mutaciones codificantes.
La proteína RET es un receptor tirosina quinasa que parece transducir las señales de crecimiento y diferenciación en varios tejidos en desarrollo, incluidos los derivados de la cresta neural. El factor neurotrófico derivado de la línea celular glial (GDNF) y la neurturina se han identificado como ligandos para RET, que es esencial para el desarrollo normal del sistema nervioso entérico y necesario para la activación de RET. Se han identificado mutaciones tanto en GDNF como en neurturina en pacientes con enfermedad de Hirschsprung. Los modelos de ratón han demostrado que la RET es necesaria para la migración, supervivencia, proliferación y diferenciación de las células derivadas de la cresta neural que dan lugar al sistema nervioso entérico, y se ha demostrado que el grado de aganglionosis es proporcional a la concentración de RET.
La fuerte asociación entre la trisomía 21 (síndrome de Down) y la enfermedad de Hirschsprung puede explicarse en parte por las mutaciones en el gen de la molécula de adhesión celular del síndrome de Down (DSCAM). En individuos con enfermedad de Hirschsprung asociada al síndrome de Down, las mutaciones en el gen DSCAM parecen producir una transmisión excesiva de un alelo RET hipomórfico, lo que explica la predisposición a enfermedad de Hirschsprung. La misma asociación se encontró para algunos casos de enfermedad de Hirschsprung sin síndrome de Down.
Síndromes asociados
La enfermedad de Hirschsprung se asocia con anomalías cromosómicas, especialmente el síndrome de Down y varios otros síndromes monogénicos:
- Trisomía 21 (síndrome de Down)
- Síndrome de Bardet-Biedl (BBS)
- Hipoplasia cartilaginosa-capilar.
- Síndrome de hipoventilación central congénita (CCHS)
- Disautonomía familiar.
- Neoplasia endocrina múltiple tipo 2 (MEN2).
- Síndrome de Mowat-Wilson (MWS)
- Síndrome de Smith-Lemli-Opitz
- Síndrome de Waardenburg
Para la trisomía 21, CCHS y BBS, RET actúa como un gen modificador para el fenotipo Hirschsprung. La evaluación realizada por un genetista clínico es valiosa para todos los pacientes con características o anomalías sindrómicas, y también para aquellos sin anomalías aparentes asociadas.
Otras anomalías congénitas
Aproximadamente del 20 al 25% de los pacientes con enfermedad de Hirschsprung tienen anomalías congénitas asociadas, a menudo, aunque no siempre, en asociación con uno de los síndromes descritos anteriormente.
Anomalías genitourinarias
Las anomalías congénitas del riñón y del tracto urinario (CAKUT), que incluyen hidronefrosis e hipoplasia renal, son particularmente comunes. Esta asociación no se explica por una relación simple con RET o GDNF, pero estos genes podrían estar involucrados como modificadores de la enfermedad. En un informe de 106 pacientes con enfermedad de Hirschsprung que se sometieron a un examen ecográfico de rutina, se encontró CAKUT en aproximadamente el 20% de los individuos con enfermedad de Hirschsprung no sindrómica y en el 40% de los que tenían enfermedad sindrómica. Debido a esta alta frecuencia, los autores de dicho informe sugirieron que estos bebés fueran sometidos a exámenes ecográficos de rutina para detectar malformaciones del sistema urinario.
Discapacidad visual y auditiva
En la serie descrita anteriormente, se encontraron anomalías oftalmológicas en aproximadamente el 40% de los individuos con enfermedad de Hirschsprung. La mayoría eran errores refractivos (hipermetropía, astigmatismo o miopía), pero la discapacidad visual estaba presente en el 9.4%. La discapacidad auditiva se encontró en aproximadamente el 5% de las personas con enfermedad de Hirschsprung, aproximadamente tres veces la tasa de la población general. Los autores sugieren pruebas de detección de rutina para el deterioro de la audición, utilizando protocolos para bebés con mayor riesgo.
Cardiopatía congénita
La cardiopatía congénita se encuentra en aproximadamente el 50% de las personas con enfermedad de Hirschsprung sindrómica (generalmente síndrome de Down), pero es inusual en pacientes sin un síndrome asociado.
Malformaciones anorrectales
La enfermedad de Hirschsprung también puede ocurrir en asociación con malformaciones anorrectales (ARM). La posibilidad de enfermedad de Hirschsprung debe considerarse en pacientes con ARM que desarrollan estreñimiento que no responde al tratamiento estándar, y en aquellos con otros síntomas que sugieren enfermedad de Hirschsprung.
Así se presenta tu paciente
La mayoría de los pacientes con enfermedad de Hirschsprung se diagnostican en el período neonatal. Los pacientes se presentan con síntomas de obstrucción intestinal distal: emesis biliar, distensión abdominal y falta de expulsión de meconio o heces. El diagnóstico puede ser sugerido por un retraso en el paso del primer meconio (más de 48 horas). A las 48 horas de vida, el 100% de los recién nacidos a término normal pasarán meconio. En contraste, entre el 45 y el 90% de los bebés con enfermedad de Hirschsprung no podrán pasar meconio en dicho periodo. Sin embargo, el paso de las heces dentro de los primeros dos días de vida no excluye el diagnóstico. Puede haber una expulsión explosiva de gases y heces después del examen rectal digital (signo del chorro o explosivo), que puede aliviar la obstrucción temporalmente.
Los bebés afectados también pueden presentar inicialmente una enterocolitis, una enfermedad potencialmente mortal en la que los pacientes tienen una imagen similar a la sepsis con fiebre, vómitos, diarrea y distensión abdominal, que puede progresar a megacolon tóxico. Los pacientes con enterocolitis requieren restitución de líquidos, tratamiento con antibióticos por vía intravenosa, que incluye cobertura para bacterias anaeróbicas, irrigaciones rectales y, en casos raros, una colostomía de urgencia. Una complicación rara de la enfermedad de Hirschsprung es el vólvulo, que puede afectar al sigmoide y, con menos frecuencia, al colon transverso y al ciego.
Los pacientes con enfermedad menos grave (generalmente porque se afecta un segmento más corto del colon) pueden no ser diagnosticados de manera temprana. En aproximadamente el 10% de los individuos, la enfermedad de Hirschsprung se diagnostica después de los tres años de edad. Estos pacientes suelen tener antecedentes de estreñimiento crónico y falta de crecimiento. Aunque es poco común, la enfermedad de Hirschsprung puede ser diagnosticada hasta la edad adulta. Los pacientes presentan síntomas de distensión abdominal y un largo antecedente de estreñimiento refractario sin incontinencia fecal. Algunos de estos pacientes pueden tener enfermedad de Hirschsprung de “segmento ultracorto”.
Evaluación
Debe sospecharse la enfermedad de Hirschsprung en pacientes con los síntomas clínicos mencionados anteriormente. Un alto índice de sospecha es apropiado para los bebés con una condición predisponente como el síndrome de Down o para aquellos con antecedentes familiares de enfermedad de Hirschsprung.
Probable enfermedad de Hirschsprung
Las siguientes características en los bebés sugieren una alta sospecha de enfermedad de Hirschsprung y justifican una evaluación completa urgente, que generalmente consiste en un enema de contraste y una biopsia rectal por succión:
- Síntomas de obstrucción, incluyendo emesis biliosa, distensión abdominal y falta de evacuación de las heces.
- No pasar meconio dentro de las 48 horas posteriores al nacimiento.
- Estreñimiento y trisomía 21 (síndrome de Down) u otra afección que se sepa que está asociada con la enfermedad de Hirschsprung o antecedente familiar de enfermedad de Hirschsprung.
- Estreñimiento y examen físico que sugieren enfermedad de Hirschsprung (distensión abdominal, esfínter anal apretado o signo del chorro en el examen digital).
Se justifica un nivel moderado de sospecha de enfermedad de Hirschsprung para los neonatos con un retraso moderado bien documentado en el paso de meconio (> 48 horas pero <72 horas) pero sin otros síntomas (sin distensión abdominal, vómitos o problemas de alimentación). La práctica varía con respecto al manejo de estos bebés. Deben someterse a un examen físico cuidadoso y a la exclusión de otras causas de retraso en el paso del meconio, incluidas las malformaciones anorrectales.
También deben observarse y evaluarse rápidamente para detectar la enfermedad de Hirschsprung si desarrollan síntomas de estreñimiento o distensión abdominal. También sería razonable realizar un enema de contraste y una biopsia rectal por succión en estos bebés, especialmente si no se puede asegurar una observación cercana. La evaluación urgente es esencial si el bebé desarrolla síntomas de obstrucción o enterocolitis.
Probable enterocolitis
Cualquier neonato con síntomas de fiebre, vómitos, distensión abdominal y diarrea explosiva, debe ser evaluado con urgencia para detectar enterocolitis asociada a Hirschsprung (HAEC). Otros síntomas incluyen letargo u obstipación. La enterocolitis asociada a Hirschsprung rara vez ocurre en neonatos, excepto cuando el diagnóstico de enfermedad de Hirschsprung se pasa por alto o se retrasa. Todos estos pacientes deben someterse a un examen rectal, realizado con un dedo (digital) o con un dilatador anal de diámetro pequeño; una liberación explosiva de gas durante este examen apoya el diagnóstico.
La evaluación también debe incluir una radiografía abdominal. El diagnóstico de HAEC está justificado por signos de íleo, incluidos niveles hidroaéreos y un intestino dilatado. No se debe realizar un enema de contraste si se sospecha de HAEC debido al riesgo de perforación intestinal.
Estreñimiento refractario crónico
Para bebés mayores y niños pequeños con estreñimiento refractario crónico (de seis meses a tres años), el nivel de sospecha de enfermedad de Hirschsprung se guía por la exploración física. Para aquellos con problemas de desarrollo y otros signos que sugieren enfermedad de Hirschsprung en la exploración física (distensión abdominal, ausencia de heces en la ampolla rectal, esfínter anal apretado o signo del chorro en el examen), se justifica un nivel moderado de sospecha de enfermedad de Hirschsprung.
Estos pacientes deben ser evaluados para enfermedad de Hirschsprung, pero el tiempo y la secuencia de las pruebas de diagnóstico es electivo. En este grupo, la manometría anorrectal es una excelente prueba de detección si está disponible. Un reflejo inhibidor anorectal normal excluye la enfermedad de Hirschsprung. Para aquellos sin síntomas o signos que sugieran enfermedad de Hirschsprung, se justifica un menor nivel de sospecha de enfermedad de Hirschsprung. Para tales pacientes, es razonable evaluar con una radiografía simple y basar la indicación de pruebas en los resultados.
Pruebas Diagnósticas
La biopsia rectal se considera el estándar de oro para el diagnóstico y puede apoyarse en los hallazgos en las radiografías abdominales, enema baritado o manometría anorrectal. Cada uno de estos procedimientos tiene ventajas y desventajas relacionadas con la disponibilidad, la experiencia técnica, la exposición a la radiación y la invasividad. Los pasos diagnósticos dependen del nivel de sospecha de enfermedad de Hirschsprung, si existe preocupación acerca de la enterocolitis por Hirschsprung (que requiere manejo de urgencia) y de los recursos disponibles y las preferencias institucionales o clínicas.
Se recomienda un enema baritado en lugar de biopsia por succión como procedimiento de diagnóstico inicial. Si se ve una zona de transición clara, el estudio es virtualmente patognomónico de la enfermedad de Hirschsprung y ayuda al cirujano a planificar la operación. Si no se ve una zona de transición, la enfermedad de Hirschsprung no se puede excluir por completo. Siempre se debe confirmar el diagnóstico mediante biopsia, incluso cuando el enema baritado muestra características típicas de la enfermedad de Hirschsprung.
Algunos médicos comienzan la evaluación con manometría anorrectal, seguida de una biopsia rectal por succión si los hallazgos son anormales. Si se observan ganglios en una biopsia por succión realizada correctamente, se excluye la enfermedad de Hirschsprung. La ausencia de ganglios en la biopsia por succión sugiere fuertemente la enfermedad de Hirschsprung, si la muestra es adecuada.
Enema baritado
En los bebés con sospecha de enfermedad de Hirschsprung, un enema baritado puede apoyar el diagnóstico de enfermedad de Hirschsprung; se realiza sin preparar el recto. Sin embargo, esta prueba no es suficiente para excluir el diagnóstico de enfermedad de Hirschsprung, especialmente en recién nacidos u otros individuos con una alta sospecha clínica de la enfermedad. Un enema baritado también es útil para la planificación prequirúrgica porque puede ayudar al cirujano a localizar la zona de transición y determinar la longitud del segmento agangliónico, aunque la ubicación de la zona de transición en la radiografía no siempre es compatible con su verdadera ubicación patológica.
La presencia de una “zona de transición”, que representa el cambio desde el calibre normal del recto y segmento agangliónico hacia el colon dilatado proximal, es virtualmente patognomónica de la enfermedad de Hirschsprung. La zona de transición usualmente se encuentra en la región rectosigmoidea, y se observa mejor en las vistas lateral y oblicua. En pacientes con afectación colónica total, todo el colon puede parecer normal, pero pueden verse asas dilatadas del intestino delgado distal.
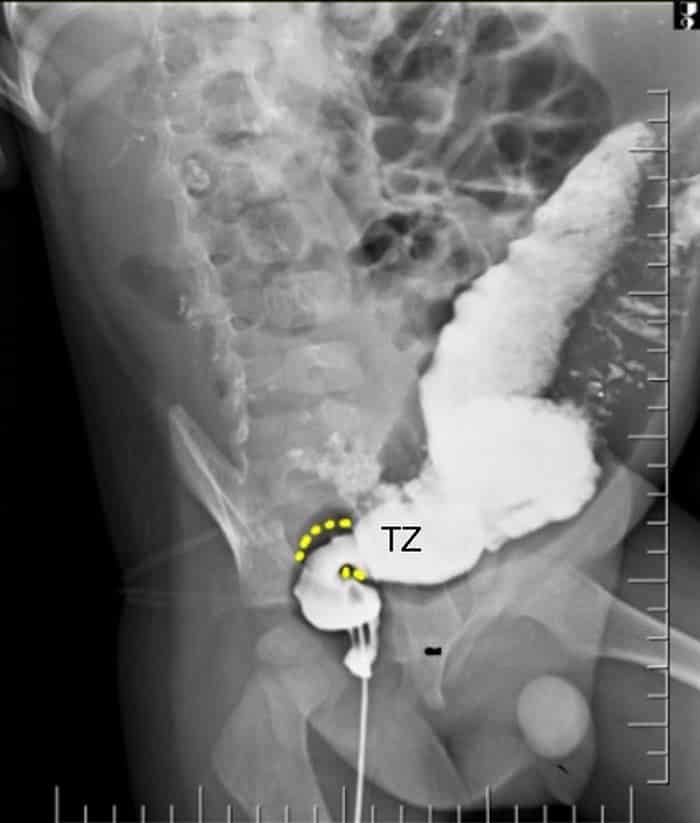
Si no se detecta claramente una zona de transición, una radiografía de seguimiento posterior a la evacuación 24 horas después puede revelar un contraste residual retenido en el colon, lo que sugiere el diagnóstico. El índice rectosigmoideo, es decir, la proporción entre el diámetro del recto y el colon sigmoide, es típicamente > 1 en niños sin enfermedad de Hirschsprung. La inversión de esta relación, aunque se observa con menos frecuencia que en una zona de transición, es un signo útil de la enfermedad de Hirschsprung en bebés y niños mayores.
Manometría anorrectal
La manometría anorrectal a veces se usa para ayudar en el diagnóstico y es especialmente útil en pacientes con enfermedad del segmento ultracorta. Es útil como prueba de detección porque un estudio claramente normal que demuestra la relajación del esfínter anal interno con distensión del recto excluye el diagnóstico de enfermedad de Hirschsprung. La falta de relajación del esfínter anal interno con distensión rectal del globo sugiere enfermedad de Hirschsprung. La manometría anorrectal tiene un valor predictivo positivo que se reporta que es del 75 al 95%, pero es menos precisa en los bebés menores de un mes de edad y en aquellos con estreñimiento crónico de larga duración.
Radiografía abdominal
Una radiografía simple de abdomen no es útil para excluir el diagnóstico de enfermedad de Hirschsprung, excepto quizás en pacientes en los que existe una baja sospecha de enfermedad (por ejemplo, niños con estreñimiento refractario moderado y examen anorectal normal). Si se realiza una radiografía simple, la posibilidad de enfermedad de Hirschsprung es sugerida por signos de obstrucción intestinal distal, es decir, disminución o ausencia de aire en el recto y asas intestinales dilatadas proximales a la región agangliónica. En ocasiones, una revisión cuidadosa de las radiografías simples puede revelar la zona de transición incluso cuando no es visible en el enema de contraste.
Diagnóstico de la Enfermedad de Hirschsprung
El diagnóstico de la enfermedad de Hirschsprung se basa en las características clínicas descritas anteriormente, generalmente respaldadas por enema baritado o manometría anorrectal. El diagnóstico se establece mediante biopsia rectal.
Biopsia rectal
La biopsia rectal por succión se puede realizar al lado de la cama o en un entorno ambulatorio sin necesidad de anestesia general. Se debe tomar una biopsia a 2 cm por arriba del nivel de la línea dentada para evitar la zona de 1 a 2 cm de aganglionosis fisiológica que normalmente está presente. Se debe tomar una segunda biopsia proximal a la primera. Generalmente se obtiene tejido adecuado para su análisis en la mayoría de los pacientes. Se pueden realizar biopsias por succión repetidas o biopsias de espesor total bajo anestesia general si la biopsia inicial no es diagnóstica (es decir, si se obtiene un tejido insuficiente).
El diagnóstico de enfermedad de Hirschsprung se establece si las células ganglionares están ausentes en la biopsia rectal, siempre que la muestra de tejido sea adecuada. Los hallazgos que apoyan el diagnóstico incluyen la presencia de fibras nerviosas hipertróficas, aumento de la actividad de acetilcolinesterasa y disminución o ausencia de fibras inmunoreactivas a calretinina en la lámina propia. Las fibras nerviosas excesivamente engrosadas pueden no aparecer hasta después de las ocho semanas de edad.
Una biopsia rectal normal prácticamente excluye la enfermedad de Hirschsprung, siempre y cuando las muestras de biopsia se obtengan del sitio correcto y contengan al menos una pequeña cantidad de muscular de la mucosa. Por tanto, una biopsia rectal por succión es más sensible y específica que el enema baritado y la manometría anorrectal para el diagnóstico de la enfermedad de Hirschsprung en niños de hasta tres años.
Diagnóstico diferencial
Otros trastornos que pueden presentarse con obstrucción intestinal en un recién nacido incluyen:
- Malformaciones gastrointestinales, como atresia intestinal, quistes de duplicación o malrotación.
- Íleo meconial por fibrosis quística.
- Trastornos asociados con ganglioneuromatosis, incluyendo neoplasia endocrina múltiple 2B.
- Trastornos que causan seudoobstrucción intestinal crónica, incluyendo displasia neuronal intestinal.
- El síndrome del tapón meconial.
- El síndrome del colon izquierdo pequeño.
Esta última entidad generalmente se presenta en bebés de madres diabéticas y parece deberse a una dismotilidad transitoria del colon izquierdo, lo que lleva a un retraso en el paso de las heces. Un enema baritado hace el diagnóstico, mostrando un colon izquierdo contrastado, y el problema generalmente remite después de unos días. Estos neonatos deben someterse a una biopsia rectal para asegurarse de que no tienen enfermedad de Hirschsprung.
En bebés mayores y niños, la principal consideración en el diagnóstico diferencial es el estreñimiento funcional. Otras posibilidades incluyen anomalías anorrectales, acalasia del esfínter anal interno, hipotiroidismo y seudoobstrucción intestinal crónica. La enfermedad de Hirschsprung clásica también debe distinguirse de la de segmento ultracorto, como se explica a continuación.
Enfermedad de Hirschsprung de Segmento Ultracorto
El término “enfermedad de Hirschsprung de segmento ultracorto” se usa a veces para describir una variante caracterizada por un segmento muy corto de aganglionosis que se extiende de 2 a 4 cm. proximal al esfínter anal interno. La mayoría de los expertos están de acuerdo en que existe esta variante, aunque hay cierta controversia sobre este punto. El cuadro clínico es similar a la variante clásica de segmento corto (que afecta a la mayor parte o todo el recto y parte del colon sigmoide), excepto que el grado de estreñimiento puede ser menos grave y las complicaciones del retraso del crecimiento y la enterocolitis son menos probables.
En el enema baritado, el recto puede dilatarse hasta el esfínter interno y puede que no haya una zona de transición visible. Si se realiza una manometría anorrectal, el reflejo inhibitorio anorrectal está ausente, como ocurre en las otras formas de enfermedad de Hirschsprung. La falta de relajación anorrectal es la base fisiológica de las características clínicas. El diagnóstico de enfermedad de Hirschsprung de segmento ultracorto se establece mediante la toma de dos biopsias:
- Una biopsia tomada cerca de la línea dentada que muestra aganglionosis: esto distingue a la enfermedad de Hirschsprung de segmento ultracorto de la acalasia interna del esfínter anal (que tiene hallazgos similares en la manometría anorrectal, pero en la acalasia hay células ganglionares presentes).
- Una biopsia tomada aproximadamente a 4 cm por arriba del esfínter interno que muestra células ganglionares normales: esta biopsia distingue a la enfermedad de Hirschsprung de segmento ultracorto de la variante clásica, en la cual no habrá células ganglionares.
Es importante distinguir a los pacientes con enfermedad de Hirschsprung de segmento ultracorto de aquellos con la variante clásica porque los primeros pueden no requerir de cirugía. Algunos pacientes responden al tratamiento intestinal con dieta, ablandadores de heces y laxantes. Otros responden a las inyecciones de toxina botulínica. Si estas medidas fallan, se debe considerar una miomectomía, retirando una tira de 0.5 a 1.0 cm de ancho de músculo liso circular en la línea media posterior, desde el nivel del esfínter anal interno hasta el intestino ganglionar normal.
Tratamiento de la Enfermedad de Hirschsprung
El pilar del tratamiento es la cirugía. Los objetivos son resecar el segmento afectado del colon, aproximar el intestino ganglionar normal al ano y preservar la función interna del esfínter anal. Se han desarrollado muchas técnicas quirúrgicas. La elección entre ellas generalmente se basa en la preferencia del cirujano, ya que las tasas generales de complicaciones y los resultados a largo plazo son similares. La técnica tradicional fue una extracción abdominoperineal en dos o tres etapas, en la que los pacientes inicialmente se sometieron a una colostomía (para permitir que el intestino dilatado se descomprima) con una reparación definitiva realizada más tarde.
Sin embargo, la mayoría de los centros ahora realizan el procedimiento en una etapa, un enfoque que no parece aumentar las tasas de complicaciones. Las reparaciones con asistencia laparoscópica y transanal son comunes, y ahora se prefieren a los procedimientos abiertos en la mayoría de los centros. Los resultados parecen ser iguales a la extracción abdominoperineal tradicional con el beneficio adicional de la reanudación temprana de la alimentación enteral, menos dolor, menor tiempo de hospitalización y cicatrices menos visibles.
Debes estar alerta a los signos o síntomas de anomalías congénitas en pacientes con probable enfermedad de Hirschsprung. En todos los pacientes con enfermedad de Hirschsprung, y particularmente aquellos con enfermedad sindrómica, son comunes las anomalías genitourinarias, la discapacidad auditiva y visual. La evaluación realizada por un genetista clínico es valiosa para todos los pacientes con características o anomalías sindrómicas, y también para aquellos sin anomalías aparentes asociadas. Los pacientes con enfermedad de Hirschsprung de segmento ultracorto pueden no requerir cirugía.
Referencias Bibliográficas
Best KE, Addor MC, Arriola L, et al. Hirschsprung’s disease prevalence in Europe: a register based study. Birth Defects Res A Clin Mol Teratol 2014; 100:695.
Ieiri S, Suita S, Nakatsuji T, et al. Total colonic aganglionosis with or without small bowel involvement: a 30-year retrospective nationwide survey in Japan. J Pediatr Surg 2008; 43:2226.
Fu M, Tam PK, Sham MH, Lui VC. Embryonic development of the ganglion plexuses and the concentric layer structure of human gut: a topographical study. Anat Embryol (Berl) 2004; 208:33.
McKeown SJ, Stamp L, Hao MM, Young HM. Hirschsprung disease: a developmental disorder of the enteric nervous system. Wiley Interdiscip Rev Dev Biol 2013; 2:113.
Goldstein AM, Hofstra RM, Burns AJ. Building a brain in the gut: development of the enteric nervous system. Clin Genet 2013; 83:307.
Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet 2008; 45:1.
Parisi MA, Kapur RP. Genetics of Hirschsprung disease. Curr Opin Pediatr 2000; 12:610.
Flori E, Girodon E, Samama B, et al. Trisomy 7 mosaicism, maternal uniparental heterodisomy 7 and Hirschsprung’s disease in a child with Silver-Russell syndrome. Eur J Hum Genet 2005; 13:1013.
Mueller C, Patel S, Irons M, et al. Normal cognition and behavior in a Smith-Lemli-Opitz syndrome patient who presented with Hirschsprung disease. Am J Med Genet A 2003; 123A:100.
de Pontual L, Pelet A, Clement-Ziza M, et al. Epistatic interactions with a common hypomorphic RET allele in syndromic Hirschsprung disease. Hum Mutat 2007; 28:790.
Sarioglu A, Tanyel FC, Büyükpamukçu N, Hiçsönmez A. Hirschsprung-associated congenital anomalies. Eur J Pediatr Surg 1997; 7:331.
Pini Prato A, Musso M, Ceccherini I, et al. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine (Baltimore) 2009; 88:83.
Hofmann AD, Puri P. Association of Hirschsprung’s disease and anorectal malformation: a systematic review. Pediatr Surg Int 2013; 29:913.
Khan AR, Vujanic GM, Huddart S. The constipated child: how likely is Hirschsprung’s disease? Pediatr Surg Int 2003; 19:439.
Janssen Lok M, Rassouli-Kirchmeier R, Köster N, et al. Development of Nerve Fibre Diameter in Young Infants With Hirschsprung Disease. J Pediatr Gastroenterol Nutr 2018; 66:253.
Mao YZ, Tang ST, Li S. Duhamel operation vs. transanal endorectal pull-through procedure for Hirschsprung disease: A systematic review and meta-analysis. J Pediatr Surg 2018; 53:1710.
Teitelbaum DH, Cilley RE, Sherman NJ, et al. A decade of experience with the primary pull-through for hirschsprung disease in the newborn period: a multicenter analysis of outcomes. Ann Surg 2000; 232:372.
Ramesh JC, Ramanujam TM, Yik YI, Goh DW. Management of Hirschsprung’s disease with reference to one-stage pull-through without colostomy. J Pediatr Surg 1999; 34:1691.
Sulkowski JP, Cooper JN, Congeni A, et al. Single-stage versus multi-stage pull-through for Hirschsprung’s disease: practice trends and outcomes in infants. J Pediatr Surg 2014; 49:1619.
Bonnard A, de Lagausie P, Leclair MD, et al. Definitive treatment of extended Hirschsprung’s disease or total colonic form. Surg Endosc 2001; 15:1301.
Hackam DJ, Filler RM, Pearl RH. Enterocolitis after the surgical treatment of Hirschsprung’s disease: risk factors and financial impact. J Pediatr Surg 1998; 33:830.
Cheng S, Wang J, Pan W, et al. Pathologically assessed grade of Hirschsprung-associated enterocolitis in resected colon in children with Hirschsprung’s disease predicts postoperative bowel function. J Pediatr Surg 2017; 52:1776.
Pini Prato A, Gentilino V, Giunta C, et al. Hirschsprung disease: do risk factors of poor surgical outcome exist? J Pediatr Surg 2008; 43:612.
Anupama B, Zheng S, Xiao X. Ten-year experience in the management of total colonic aganglionosis. J Pediatr Surg 2007; 42:1671.
Pontarelli EM, Ford HR, Gayer CP. Recent developments in Hirschsprung’s-associated enterocolitis. Curr Gastroenterol Rep 2013; 15:340.
Aworanti OM, McDowell DT, Martin IM, Quinn F. Does Functional Outcome Improve with Time Postsurgery for Hirschsprung Disease? Eur J Pediatr Surg 2016; 26:192.
Stensrud KJ, Emblem R, Bjørnland K. Anal endosonography and bowel function in patients undergoing different types of endorectal pull-through procedures for Hirschsprung disease. J Pediatr Surg 2015; 50:1341.
Bischoff A, Frischer J, Knod JL, et al. Damaged anal canal as a cause of fecal incontinence after surgical repair for Hirschsprung disease – a preventable and under-reported complication. J Pediatr Surg 2017; 52:549.




