Síndrome Urémico Hemolítico y Púrpura Trombótica Trombocitopénica.
Estos padecimientos se caracterizan por daño renal agudo, anemia hemolítica microangiopática y trombocitopenia. El síndrome urémico hemolítico es más frecuente en pacientes pediátricos, mientras que la púrpura trombótica trombocitopénica lo es en adultos. Ambas se conocen como microangiopatía trombótica (MAT). Revisemos lo esencial de este tema para tu práctica clínica.
En la microangiopatía trombótica se genera lesión endotelial inflamatoria, acompañada de consumo del complemento y alteración en la fragmentación del factor de von Willebrand.
Síndrome Urémico Hemolítico
El síndrome urémico hemolítico típico se presenta en niños y es causado por las toxinas Shiga de Shigella dysenteriae, así como algunos serotipos de E. colli, como el O157:H7 y O104:H4. Las toxinas Shiga causan daño directo a las células epiteliales del riñón (podocitos y células tubulares), células mesangiales y endoteliales vasculares. Casi todos los casos son esporádicos, presentándose brotes en deficiencias sanitarias locales. El síndrome urémico hemolítico atípico, por el contrario, se presenta en adultos y es secundario a una activación descontrolada del complemento en membranas celulares; incluyendo al endotelio vascular y células renales.
Ello se debe a una alteración genética de las proteínas reguladoras de la activación del complemento; tales como: el factor H (CFH), las relacionadas al factor H (CFHRs), CFI y MCP o a anormalidad de las proteínas que aceleran esta vía como el CFB y C3. Cabe mencionar que la deficiencia del factor H (CFH) o I (CFI) también puede ser adquirida, secundaria a autoanticuerpos que inhiben su actividad.
Púrpura Trombótica Trombocitopénica
En el caso de la púrpura trombótica trombocitopénica los multímeros del factor de von Willebrand no se pueden fraccionar, o lo hacen sólo parcialmente, debido a la deficiencia grave en la actividad del factor ADAMTS13 (menor al 10%). Esto produce una agregación plaquetaria anormal en las células endoteliales. Sin embargo, el diagnóstico es clínico, dado que la determinación de la actividad de ADAMTS13 no es posible hasta después de varios días. Dicha deficiencia puede ser hereditaria, es decir, el síndrome de Upshaw-Schulman, o adquirida por inhibición de su actividad causada por autoanticuerpos.
La púrpura trombótica trombocitopénica se caracteriza, dentro de los síndromes de microangiopatía trombótica, por escasa afectación de la función renal. Ello a pesar de que se observan microtrombos en el riñón. Sin embargo, la púrpura trombótica trombocitopénica ocasiona mayores manifestaciones sistémicas que los otros síndromes primarios de microangiopatía trombótica. Puede haber afectación del sistema nervioso central, corazón, páncreas, tiroides, glándulas adrenales y mucosa intestinal. Los pulmones por lo general se ven respetados.
| Causa | Mecanísmo de la lesión |
|---|---|
| SHU típico | Infección por Toxina Shiga (E. coli o Shigella). |
| SHU atípico | Defecto congénito o adquirido del complemento y actividad anormal. |
| PTT | Déficit de la actividad de ADAMTS13 congénito o adquirido. |
| Infecciones | Shock séptico, seroconversión en el VIH, influenza H1N1 |
| Fármacos | Ticlopidina, quinina, tacrolimus, clopidogrel, ciclosporina, bleomicina, mitomicina, cisplatino, rifampicina, gemcitabina. |
| Neoplasias | Obstrucción de pequeño vaso por elementos tumorales. |
| Enfermedades del tejido conectivo: LES, esclerodermia, crioglobulinemia, Sx. antifosfolípidos. | Anticuerpos dependientes del complemento, citotóxicos para el endotelio. |
| Preeclampsia y HELLP | Disfunción endotelial por factor placentario. |
| Rechazo de transplante | Autoinmune |
| Déficit del metabolismo de la cobalamina | |
| Hemoglobinuria paroxística nocturna |
Bajo el Microscopio
La lesión en la microangiopatía trombótica cursa con engrosamiento de la íntima arteriolar, grado variable de hipertrofia muscular y necrosis de la pared en algunos casos; a menudo con trombos fibrinoides intraluminales. Los vasos principalmente afectados son las pequeñas arterias renales y las arteriolas aferentes. Las arterias interlobulillares muestran engrosamiento de la íntima con disminución de la luz. Los glomérulos presentan un engrosamiento uniforme de las paredes capilares y necrosis, así como leve proliferación celular.
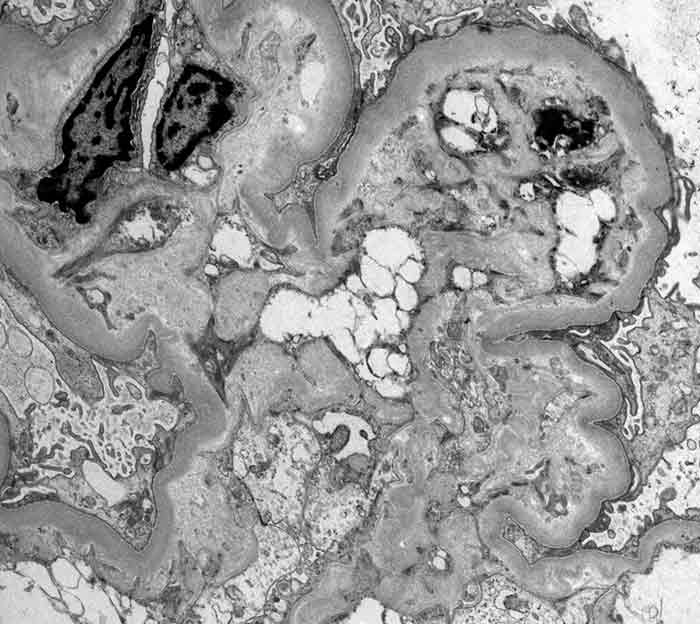
A la microscopía electrónica podrás observar engrosamiento de la membrana basal, rarefacciones subendoteliales y, en ocasiones, un doble contorno de la pared capilar. Con inmunofluorescencia se podrá detectar fibrinógeno en las paredes y luz vasculares, al igual que depósito de complemento.
Así se presenta tu paciente
Los pacientes con microangiopatía trombótica se presentan con oliguria o anuria, siendo más grave en el síndrome urémico hemolítico y requiriendo en ocasiones de diálisis. Hay presencia de hematuria y proteinuria de hasta 3 gr./día, en ocasiones cilindros hialinos y granulosos. Es frecuente la hipertensión grave o incluso maligna por la activación del sistema renina-angiotensina-aldosterona. La púrpura puede ir acompañada de fiebre, siendo más rara en el síndrome urémico hemolítico. La afectación neurológica puede presentarse en ambos padecimientos, siendo mucho más frecuente en la púrpura trombótica trombocitopénica.
Puede cursar con confusión, desorientación, convulsiones y coma. Los pacientes con púrpura trombótica trombocitopénica y afectación neurológica tienen el peor pronóstico. La trombocitopenia por consumo está presente en ambos padecimientos. Siendo de mayor relevancia en la púrpura trombótica trombocitopénica con valores inferiores a 30,000/ml.; mientras que en el síndrome urémico hemolítico se localiza entre los 80 y 100,000/ml. Se manifiesta mediante púrpura cutánea, hemorragias retinianas, hematuria, epistaxis, petequias, melenas, metrorragias y equimosis.
Anemia Hemolítica Microangiopática (AHM)
Se trata de una hemólisis no autoinmune o Coombs (directo) negativa. Ocurre secundaria al traumatismo de los eritrocitos al circular por vasos ocluidos con formación de esquistocitos y células casco. En consecuencia, es posible la elevación de reticulocitos, bilirrubina indirecta y LDH, así como disminución de haptoglobulinas. Cabe resaltar que no todas las anemias hemolíticas microangiopáticas son causadas por una púrpura trombótica trombocitopénica; sin embargo, casi todos los casos de púrpura cursan con anemia hemolítica microangiopática y trombocitopenia.
Claves del Abordaje Diagnóstico
Una vez que por clínica tu sospecha es de síndrome urémico hemolítico o púrpura trombótica trombocitopénica, lo primero será confirmar la presencia de anemia hemolítica microangiopática más trombocitopenia. De igual manera, tendrás que excluir diagnósticos diferenciales (ver tabla abajo) que pueden presentarse con estos hallazgos. El diagnóstico de AHM y trombocitopenia lo realizas solicitando un frotis de sangre. Si se descarta AHM, deberás considerar otros padecimientos asociados a pancitopenia.
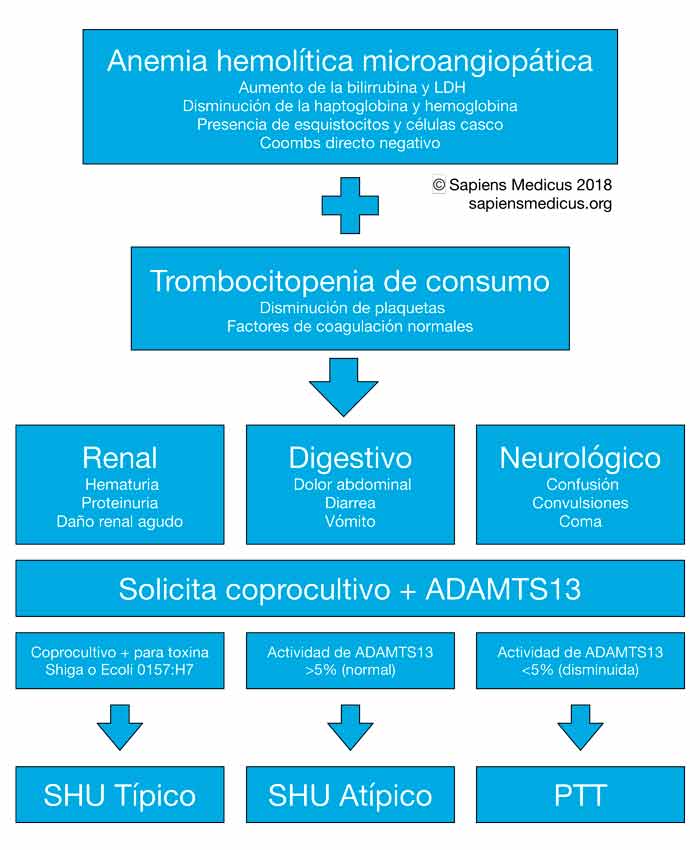
Diagnósticos Diferenciales
| Dx. diferencial | Características |
|---|---|
| Coagulación intravascular diseminada | Disminución del fibrinógeno, Aumento de TP y TTPA, Disminución de los Factores V y VIII. |
| Vasculitis de tipo ANCA | ANCA positivos. |
| Síndrome antifosfolípidos | Anticoagulante lúpico. |
| Esclerodermia agudizada | Clínica de esclerodermia Anti-anticentrómero, Anti-Scl-70 y Anti-ARN polimerasa positivos. |
| Púrpura trombótica idiopática | No cursa con anemia hemolítica, el Coombs directo es positivo si cursa con hemólisis. |
| Púrpura de Henoch-Schönlein | Artritis no erosiva, plaquetas normales, púrpura no necrotizante. |
Tratamiento: PTT o no PTT.
La decisión más importante es determinar el inicio de tratamiento con plasmaféresis por sospecha de púrpura trombótica trombocitopénica o diferir el tratamiento para descartar otros síndromes de microangiopatía trombótica. Ello debido a que un paciente con púrpura trombótica trombocitopénica sin plasmaféresis tiene una mortalidad de hasta el 90%. Por tanto, toma la decisión con base en la probabilidad del diagnóstico de púrpura trombótica trombocitopénica versus los riesgos del tratamiento con plasmaféresis.
Estos incluyen complicaciones asociadas al catéter venoso central, como hemorragia, infecciones o trombosis, así como reacciones secundarias al plasma. Si recibes un paciente (adulto) refiriendo astenia y adinamia, así como malestar general por varios días y se determina anemia grave, trombocitopenia, esquistocitos y escaso daño renal, sospecha de púrpura trombótica trombocitopénica e indica plasmaféresis urgente.
Tratamiento en Diagnósticos Diferenciales
Si el paciente refiere que inició con náusea y anuria, horas posterior a un compuesto de quinina, algún fármaco IV., o desarrolla hipertensión y falla renal aguda, semanas posterior al tratamiento con un inhibidor de la calcineurina o quimioterapia, sospecha de una microangiopatía trombótica secundaria a fármacos. En cuyo caso se podrá evitar la plasmaféresis.
Por último, si se trata de una paciente puérpera, con rápido desarrollo de falla renal aguda y requerimiento de diálisis, sospecha de microangiopatía trombótica del puerperio asociada al complemento. Indica tratamiento anti-complemento sin plasmaféresis. En los demás síndromes de la microangiopatía trombótica no está indicada la plasmaféresis, con excepción de los casos donde exista pobre o nula respuesta al tratamiento de soporte.
Por si pensabas indicar…
Se consideran infectivos los antiagregantes plaquetarios, como el ácido acetilsalicílico o el dipiridamol, así como la esplenectomía. La transfusión plaquetaria estará reservada únicamente cuando esté en peligro la vida del paciente. El rituximab estará indicado ante la falla del tratamiento, así como casos graves y/o alteración neurológica muy importante. El síndrome urémico hemolítico atípico es tratado con eculizumab, el cual inhibe a C5 bloqueando la activación del complejo de ataque a la membrana (CAM). Esta variante del síndrome urémico hemolítico puede recurrir, en especial en los casos secundarios a alteraciones del factor H o I.
Si tu paciente cursa con falla renal aguda por más de cuatro semanas a 3 meses puede requerir de diálisis o incluso trasplante. Por último, la mortalidad alcanza el 15% en niños y 30% en adultos. Los factores de mal pronóstico incluyen edad mayor a 40 años, fiebre por arriba de 38.5ºC y hemoglobina menor a 9 mg./dl.
| SHU típico | SHU atípico | PTT | |
|---|---|---|---|
| Edad | Niños | Adultos | Adultos |
| Etiología | Proceso infeccioso | Vía alterna del complemento | Deficiencia de actividad de ADAMTS13 |
| Daño renal agudo | Grave | Tiende a ERC | Leve a moderado |
| Afectación neurológica | Rara | Probable | Frecuente |
| Trombocitopenia | Sí | Sí | Grave |
| Fiebre | No presenta | Sí | Sí |
| Tratamiento | Soporte y plasmaféresis en mala evolución | Eculizumab | Plasmaféresis, Rituximab, Inmunosupresión |
Referencias Bibliográficas
BRAIN MC, DACIE JV, HOURIHANE DO. Microangiopathic haemolytic anaemia: the possible role of vascular lesions in pathogenesis. Br J Haematol 1962; 8:358.
Laszik ZG, Kambham N, Silva FG. Thrombotic microangiopathies. In: Heptinstall’s Pathology of the Kidney, Jennett JC, D’Agati VD, Olson JL, et al (Eds), Lippincott Williams & Wilkins, Philadelphia 2014.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014; 371:654.
Nokes T, George JN, Vesely SK, Awab A. Pulmonary involvement in patients with thrombotic thrombocytopenic purpura. Eur J Haematol 2014; 92:156.
Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010; 5:1844.
Al-Nouri ZL, Reese JA, Terrell DR, et al. Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood 2015; 125:616.
George JN. Cobalamin C deficiency-associated thrombotic microangiopathy: uncommon or unrecognised? Lancet 2015; 386:1012.
Cornec-Le Gall E, Delmas Y, De Parscau L, et al. Adult-onset eculizumab-resistant hemolytic uremic syndrome associated with cobalamin C deficiency. Am J Kidney Dis 2014; 63:119.
Grangé S, Bekri S, Artaud-Macari E, et al. Adult-onset renal thrombotic microangiopathy and pulmonary arterial hypertension in cobalamin C deficiency. Lancet 2015; 386:1011.
Bu F, Maga T, Meyer NC, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2014; 25:55.
Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost 2017; 15:312.
McMinn JR, George JN. Evaluation of women with clinically suspected thrombotic thrombocytopenic purpura-hemolytic uremic syndrome during pregnancy. J Clin Apher 2001; 16:202.
Booth KK, Terrell DR, Vesely SK, George JN. Systemic infections mimicking thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:743.
George JN. Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia. Oncology (Williston Park) 2011; 25:908.
Song D, Wu LH, Wang FM, et al. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther 2013; 15:R12.
Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Blood 2011; 118:1452.
Chhabra N, Lee S, Sakalis EG. Cobalamin deficiency causing severe hemolytic anemia: a pernicious presentation. Am J Med 2015; 128:e5.
Dimond A, George JN, Hastings C. Severe vitamin B-12 deficiency in a child mimicking thrombotic thrombocytopenic purpura. Pediatr Blood Cancer 2009; 52:420.
Nester CM, Barbour T, de Cordoba SR, et al. Atypical aHUS: State of the art. Mol Immunol 2015; 67:31.
Go RS, Winters JL, Leung N, et al. Thrombotic Microangiopathy Care Pathway: A Consensus Statement for the Mayo Clinic Complement Alternative Pathway-Thrombotic Microangiopathy (CAP-TMA) Disease-Oriented Group. Mayo Clin Proc 2016; 91:1189.
Maki DG. Don’t eat the spinach–controlling foodborne infectious disease. N Engl J Med 2006; 355:1952.
Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2007; 18:2392.
Bendapudi PK, Li A, Hamdan A, et al. Impact of severe ADAMTS13 deficiency on clinical presentation and outcomes in patients with thrombotic microangiopathies: the experience of the Harvard TMA Research Collaborative. Br J Haematol 2015; 171:836.
Hassan S, Westwood JP, Ellis D, et al. The utility of ADAMTS13 in differentiating TTP from other acute thrombotic microangiopathies: results from the UK TTP Registry. Br J Haematol 2015; 171:830.
Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010; 115:1500.
Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol 2016; 31:15.
Amorosi EL, Ultmann JE. Thrombotic thrombocytopenic purpura: Report of 16 cases and review of the literature. Medicine (Baltimore) 1966; 45:139.
Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325:393.
McClain RS, Terrell DR, Vesely SK, George JN. Plasma exchange complications in patients treated for thrombotic thrombocytopenia purpura-hemolytic uremic syndrome: 2011 to 2014. Transfusion 2014; 54:3257.
Naik S, Mahoney DH. Successful treatment of congenital TTP with a novel approach using plasma-derived factor VIII. J Pediatr Hematol Oncol 2013; 35:551.
Peyvandi F, Mannucci PM, Valsecchi C, et al. ADAMTS13 content in plasma-derived factor VIII/von Willebrand factor concentrates. Am J Hematol 2013; 88:895.




