Síndrome de Cushing y Enfermedad de Addison: Breve Revisión.
La relación de Dante Alighieri y dos grandes en el desequilibrio hormonal podemos encontrarla al relacionar la divina comedia y la endocrinología. La enfermedad de Addison y el síndrome de Cushing representan los extremos de las alteraciones del cortisol. Ambas son un reto diagnóstico y la interpretación de los exámenes de laboratorio es complicada. Te ofrecemos una breve revisión sobre su diagnóstico y tratamiento.
Síndrome de Cushing
El síndrome de Cushing se presenta como resultado de la exposición a altas concentraciones de cortisol. El diagnóstico es un desafío, ya que debes conocer la utilidad y la correcta interpretación de las pruebas diagnósticas. La mayoría de los casos se deben al tratamiento con glucocorticoides a dosis altas (síndrome de Cushing exógeno), y en menor proporción a lesiones tumorales o síndrome de Cushing endógeno; ya sea hipofisarias (enfermedad de Cushing; 65 a 70%), suprarrenales (15 a 20%) o ectópicas (15%). A pesar de que se considera una enfermedad rara, se caracteriza por una morbilidad y mortalidad alta.
La enfermedad de Cushing es ocasionada por un adenoma hipofisario en el 90% de los casos, el cual libera grandes concentraciones de ACTH. Como ya mencionamos, el síndrome de Cushing puede ser ocasionado también por tumores ectópicos productores de grandes cantidades de ACTH o CRH. El responsable en el 50% de los casos es el carcinoma microcítico de pulmón, seguido del tumor carcinoide de pulmón, timo, intestino, páncreas u ovario. Otros tumores productores de ACTH de menor frecuencia son aquellos de los islotes pancreáticos, el carcinoma medular de tiroides y el feocromocitoma.
Los tumores suprarrenales son en un 75% adenomas y el resto carcinomas o hiperplasia nodular suprarrenal, cursando con niveles bajos de ACTH. En pacientes pediátricos la etiología más frecuente de síndrome de Cushing es suprarrenal (65%), la mayoría de estos casos es un carcinoma.
Así se presenta tu paciente
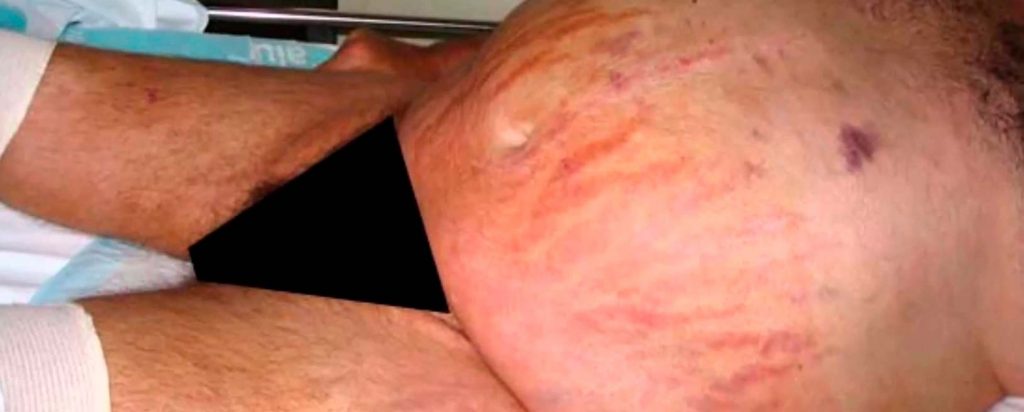
Los pacientes con síndrome de Cushing refieren ganancia de peso en el 90% de los casos, además de ciclo menstrual irregular en mujeres (85%), hirsutismo (80%), alteraciones mentales (60%) y debilidad muscular (30%). Los signos que encontraremos con mayor frecuencia son la obesidad central (97%), plétora facial (95%), cara de luna llena (90%), hipertensión arterial (75%), hematomas y fragilidad capilar (40%), estrías abdominales rojo-vino (60%), miopatía proximal (60%), edema en miembros inferiores (50%) y osteoporosis (50%).
La liberación de ACTH por un tumor ectópico ocasiona un síndrome de Cushing atípico, el cual puede ser sugerido por las alteraciones metabólicas del exceso de glucocorticoides; tal es el caso de la hiperglucemia, hipokalemia y alcalosis metabólica. Existe además hiperpigmentación (5%) y miopatía proximal. Se recomienda realizar búsqueda intencionada en aquellos pacientes con características inusuales para la edad, como una osteoporosis e hipertensión inexplicables, aquellos con datos clínicos predictivos de síndrome de Cushing, niños con talla baja, sobrepeso, y en aquellos pacientes con adenoma suprarrenal.
Estudios que serán de utilidad
Como consecuencia del efecto mineralocorticoide se genera una alcalosis hipopotasémica e hipernatremia. Recuerda que la estimulación glucocorticoide, con aumento de cortisol en plasma estimula la gluconeogénesis, con resistencia a la insulina o hiperinsulinemia. En pruebas más específicas encontraremos un valor plasmático de cortisol elevado en las primeras horas de la mañana, pérdida del ritmo circadiano con ausencia del descenso de cortisol nocturno, e inhibición de la secreción de cortisol con dosis bajas de glucocorticoides. El procedimiento para establecer el diagnóstico bioquímico y etiológico del síndrome de Cushing consiste en tres fases:
Detección Bioquímica
El test de Nugent consiste en la determinación de cortisol plasmático a las 8:00 horas tras la administración de 1 mg de dexametasona a las 23:00 horas del día anterior. En condiciones normales la cifra de cortisol es < 1.8 μg/dl. Aquellos valores > 1.8 μg/dl indican hipercortisolismo. La detección de cortisol libre urinario también nos puede orientar hacia un hipercortisolismo cuando encontramos concentraciones de cortisol > 120 μg en orina de 24 horas. En este último caso se deberán obtener muestras en dos días diferentes.
Confirmación Bioquímica
Valores de cortisol libre > 300 μg en orina de 24 horas son altamente sugerentes de síndrome de Cushing. El cortisol en plasma a las 24:00 horas > 7.5 μg/dl también es confirmatorio. Otra de las pruebas confirmatorias es la de Liddle, con ausencia de supresión de cortisol plasmático (>1.8 μg/dl) y cortisol libre urinario (>20 μg/dl) tras la administración de 0.5 mg de dexametasona cada 6 horas por 2 días. En pacientes con resultados no concluyentes se recurre a estudios de segunda línea, tales como cortisol sérico nocturno o la prueba combinada de supresión con 2 mg. de dexametasona más estímulo con CRH.
En pacientes embarazadas o con epilepsia, se realiza la determinación de cortisol libre en orina de 24 hrs. y está contraindicada la supresión con dexametasona. Por el contrario, en pacientes con enfermedad renal no es de utilidad la determinación de cortisol libre en orina de 24 hrs. y está indicada la prueba de supresión. En el síndrome de Cushing cíclico está indicado el cortisol libre urinario, mientras que en los incidentalomas suprarrenales la prueba de supresión nocturna con dexametasona es la de elección.
Identificación de la Etiología
Para saber si existe dependencia o no de ACTH determinaremos su concentración mediante el método inmunorradiométrico. Se consideran valores de ACTH < 5 pg/ml sugerentes de hipercortisolismo de origen suprarrenal, mientras que cifras superiores a 20 pg/ml sugieren un origen hipofisario o ectópico. Si los valores se encuentran entre 5 y 20 pg/ml., lo más probable es que se trate de un síndrome de Cushing ACTH dependiente; sin embargo, se realiza una prueba de estimulación con CRH para corroborarlo.
Esta última prueba se realiza administrando CRH intravenosa (no disponible en México) y determinando la ACTH y el cortisol posteriormente, esperando observar un aumento en la mayoría de pacientes con alteración hipotalámica o adenoma hipofisario productor de ACTH. Cabe resaltar que un 10% de los pacientes con enfermedad de Cushing no responde a estas pruebas. Otra prueba realizada es el protocolo de Liddle, también llamada prueba de supresión con dosis altas de dexametasona. Se administran 2 mg. de este corticoide cada 6 hrs. durante 2 días. La supresión del cortisol en orina de 24 hrs. > 50% es sugerente y > 90% altamente sugerente de enfermedad de Cushing.
Esta prueba también se puede realizar con una dosis única de 8 mg. de dexametasona. Para ello, se determinan niveles basales de cortisol a las 9 hrs. previo a la primera administración del corticoide VO., y a las 23 hrs. se vuelven a administrar otros 8 mg. VO. Al día siguiente se miden los niveles de cortisol a las 9 hrs. Una supresión > 50% con respecto al cortisol basal es sugerente de enfermedad de Cushing, mientras que un valor > 68% aumenta la especificidad de la prueba. En caso de supresión en esta prueba, está indicada la realización de una resonancia magnética para confirmar el adenoma hipofisario. Si no se observa lesión en la RM o no hay supresión se realiza un cateterismo de senos petrosos inferiores.
Cateterismo de Senos Petrosos Inferiores
Este estudio es de utilidad para el diagnóstico diferencial entre la enfermedad de Cushing y el síndrome ectópico cuando no es posible visualizar lesión en la RM con gadolinio. Los tumores carcinoides, en particular, muestran una respuesta positiva al ser estimulados por las pruebas funcionales; lo que hace difícil su diferenciación de un adenoma hipofisario. La presencia de hipokalemia y/o alcalosis metabólica indican este estudio y sugieren un origen ectópico del síndrome de Cushing. Un gradiente petroso-periférico de ACTH ≥ 3:1 posterior a la administración de CRH o desmopresina permite establecer el origen hipofisario de hipersecreción de ACTH.
Estudios de Imagen
La RM selar con gadolinio es el estudio de elección para la identificación de tumores hipofisarios; sin embargo, es importante considerar que en el 10% de las personas que son sometidas a este estudio se identifica un incidentaloma hipofisario. Se trata de tumores no funcionantes sin relevancia clínica. Por tanto, el diagnóstico imagenológico siempre debe ser correlacionado con las pruebas funcionales. La TAC de cortes finos es de elección en los casos de probable síndrome de Cushing ACTH independiente secundario a un tumor suprarrenal.
De igual manera, está indicada la TAC toracoabdominal ante la sospecha de un tumor ectópico secretor de ACTH o CRH. La RM abdominal puede ser de utilidad en la evaluación de tumores suprarrenales. El octreoscan permite la obtención de imágenes de gammagrafía posterior a la inyección de pentetreotide radiomarcado; los tumores productores de ACTH lo captarán, ya que expresan receptores para somatostatina.
Pseudocushing
Los pacientes con pseudocushing tienen ausencia de supresión del cortisol sérico posterior a la administración de dosis bajas de dexametasona, con niveles > 1.8 mcg/dl. pero menores a 7.5 mcg/dl. Deberás sospecharlo en pacientes con dicho hallazgo y obesidad grave, depresión, alcoholismo crónico y enfermedades que cursan con gran estrés físico. Además, hay ausencia de manifestaciones cutáneas y musculares. En el diagnóstico diferencial se realiza una prueba combinada con 2 mg. de dexametasona y estimulación con CRH. Los pacientes con enfermedad de Cushing tendrán respuesta a la CRH; en el pseudocushing dicha respuesta estará ausente. En pacientes alcohólicos la prueba de elección es la supresión del alcohol, con cortisol indetectable durante la noche del quinto día.
Tratamiento de la Enfermedad y Síndrome de Cushing
Ante la presencia de tumores suprarrenales el tratamiento de elección es la resección quirúrgica. Los casos con adenoma logran la curación posterior a la cirugía, debiéndose considerar la supresión del eje posterior a la intervención, así como la atrofia glandular contralateral. En el carcinoma suprarrenal, por el contrario, la mortalidad es elevada a pesar de la cirugía, con metástasis frecuente a hígado y pulmón.
Síndrome de Cushing ACTH-Dependiente
En pacientes con tumores hipofisarios productores de ACTH el tratamiento consiste en la resección quirúrgica por vía transesfenoidal. Ante la ausencia de un hallazgo transquirúrgico de lesión y paridad satisfecha, se realiza hipofisectomía subtotal. En caso de fallo de la intervención quirúrgica o paridad no satisfecha se indica radioterapia. Los tumores ectópicos también se extirpan y, ante la ausencia de hallazgo tumoral posterior a 6 meses de evaluación exhaustiva, se realiza suprarrenalectomía bilateral. Ante el fracaso del tratamiento quirúrgico se emplea mitotano o inhibidores de la síntesis de cortisol; tales como el ketoconazol, aminoglutetimida o metopirona.
En pacientes con hipercortisolismo grave previo a la cirugía o fracaso de la intervención quirúrgica en espera de tratamiento definitivo se puede indicar ketoconazol. La suprarrenalectomía bilateral se realiza en los casos de fallo del tratamiento quirúrgico o radioterapia o ante la presencia de efectos adversos farmacológicos. La tasa de curación es del 100% pero se requiere de sustitución con gluco- y mineralocorticoides; además, aumenta el riesgo de un síndrome de Nelson.
Enfermedad de Addison
La insuficiencia suprarrenal o enfermedad de Addison puede estar ocasionada por enfermedad autoinmune, hipotalámica o hipofisaria, así como por supresión del eje hipotálamo-hipófisis-adrenal por administración exógena o producción endógena de esteroides. La causa más frecuente de insuficiencia suprarrenal primaria (>70%) o enfermedad de Addison es la adrenalitis autoinmune, con destrucción de la corteza suprarrenal; lo que conduce a deficiencia de glucocorticoides, mineralocorticoides y andrógenos suprarrenales. La prevalencia es de un caso en 20,000 personas y puede asociarse a otras endocrinopatías, tales como diabetes mellitus, enfermedad tiroidea autoinmune, hipogonadismo, entre otras.
Es importante recordar que la tuberculosis es responsable de otro 10 a 20% de los casos de insuficiencia suprarrenal; mientras que en pacientes con SIDA puede ser secundaria a la infección glandular por citomegalovirus, M. avium intracelular, criptococo y el sarcoma de Kaposi.
Así se presenta tu paciente
Las manifestaciones clínicas de la insuficiencia suprarrenal pueden incluir hiperpigmentación, fatiga y debilidad progresivas, anorexia, náuseas, mialgias y artralgias. En la insuficiencia primaria hay afectación del tejido secretor de mineralocorticoides, cursando con hipoaldosteronismo, pérdida de sodio, hiperpotasemia, avidez por la sal, hipotensión ortostática y acidosis metabólica leve. El evento precipitante inicial es la autoinmunidad adrenal, en la que se producen los anticuerpos 21-hidroxilasa, principales responsables de esta enfermedad. La producción de estos anticuerpos puede dar inicio a los síntomas de manera insidiosa durante años y están presentes en más del 90% de los casos.
Uno de las primeras alteraciones es un aumento del nivel de renina plasmática, seguido del desarrollo de otras anomalías, incluyendo una disminución de la respuesta a la ACTH. En etapas avanzadas las suprarrenales ya manifiestan insuficiencia. La ausencia de cortisol provoca un aumento en la síntesis de ACTH y sus péptidos, generando la hiperpigmentación mucocutánea característica en labios, áreas de presión, pliegues cutáneos, nudillos, codos, rodillas y cicatrices del paciente. En las mujeres se pierde el vello púbico y axilar; además, puede haber calcificación del cartílago articular y del pabellón auricular.
Crisis Suprarrenal
La etapa final de la enfermedad se caracteriza por la crisis addisoniana. La causa global más frecuente es la suspensión súbita del tratamiento prolongado con corticoides a altas dosis. Sin embargo, en un paciente con insuficiencia suprarrenal ya establecida, la causa más frecuente es la presencia de situaciones de gran estrés físico, tales como enfermedad grave, sepsis, cirugía o traumatismo. La destrucción hemorrágica aguda de la glándula puede ocurrir ante una septicemia por Pseudomonas o meningococcemia en niños (Sx. de Waterhouse-Friederichsen); o tratamiento anticoagulante en el caso de adultos.
El paciente con crisis adrenal se presenta con fiebre elevada, deshidratación, náuseas, vómitos e hipotensión que puede progresar a choque. Además, los electrolitos séricos se caracterizan por hiperpotasemia, hiponatremia, hipercalcemia y acidosis metabólica. Se acompaña de hemoconcentración y elevación de la urea. La presencia de hiperpotasemia, hipotensión y acidosis hace sugerente el diagnóstico de insuficiencia suprarrenal primaria, dado que en la secundaria solo se observa hiponatremia.
Estudios que serán de utilidad
La determinación del cortisol basal habitualmente no es suficiente para el diagnóstico de insuficiencia suprarrenal. Si bien concentraciones <3 μg/dl son indicativas de insuficiencia, concentraciones >19 μg la descartan. Cuando sospechamos de insuficiencia suprarrenal primaria determinaremos los valores de ACTH, los cuales se van encontrar elevados en estos casos. En aquellos pacientes que presentan valores intermedios de cortisol basal (3-19 μg/dl) será necesario realizar un test de estimulación de hormona adrenocorticotropa (ACTH). La prueba gold standard para la insuficiencia suprarrenal primaria es la respuesta de cortisol tras la estimulación con ACTH.
Consiste en administrar 250 μg de ACTH IV antes de las 10:00 horas y posteriormente medir la respuesta del cortisol a los 30 y 60 minutos. Se considera una respuesta patológica, e indicativa de insuficiencia suprarrenal, un valor de cortisol por debajo de 18 μg/dl. y ausencia de elevación de la aldosterona. En la insuficiencia suprarrenal secundaria también hay ausencia de elevación del cortisol; sin embargo, la aldosterona si se eleva (> 5 ng/dl) en respuesta a la ACTH. Ello debido a que la pars glomerular (mineralocorticoide) de la suprarrenal no se encuentra atrofiada. En caso de no ser concluyente se realiza el test de hipoglucemia insulínica.
Test de Hipoglucemia insulínica y Metopirona
El test de Hipoglucemia insulínica, para el diagnóstico de insuficiencia suprarrenal secundaria, consiste en administrar 0.1-0.15 U/kg de insulina IV para inducir una hipoglucemia (glucosa < 40mg/dl) capaz de estimular todo el eje hipotálamo-hipófisis-suprarrenales. Se realiza por la mañana y se determina la glucosa y cortisol antes de la administración de la insulina y a los 30, 60, 90 y 120 minutos después de su administración. Una respuesta de cortisol inferior a 18 μg/dl es indicativa de insuficiencia suprarrenal.
Esta prueba está contraindicada en pacientes con cardiopatía isquémica, epilepsia o enfermedad cerebrovascular; además, debe realizarse con mucho cuidado ante la presencia de otros déficits hipofisarios. Por último, la prueba con metopirona consiste en bloquear la síntesis de cortisol. En pacientes sanos la metopirona ocasiona la elevación de 11-desoxicortisol, un metabolito previo al sitio de bloqueo del fármaco; dicha elevación está ausente en pacientes con insuficiencia suprarrenal central o secundaria.
Tratamiento de la Enfermedad de Addison
Se requiere la sustitución de glucocorticoides, tanto en la insuficiencia suprarrenal primaria como secundaria, y de mineralocorticoides únicamente en la primaria. En el caso de los glucocorticoides, se deberán administrar en dos a tres dosis diarias; la mayoría durante la mañana y el resto por la tarde, para ofrecer niveles similares a los del ciclo circadiano de secreción fisiológica de cortisol. Son de utilidad la cortisona, hidrocortisona o la prednisona, así como la administración nocturna de dexametasona. La dosis deberá ajustarse tomando en cuenta la sintomatología del paciente, como por ejemplo insomnio o irritabilidad.
Los pacientes hipertensos y diabéticos requieren de una menor dosis, mientras que los obesos o aquellos con tratamiento antiepiléptico requerirán dosis mayores. La sustitución de mineralocorticoides se realiza mediante fludrocortisona y se controla mediante la medición de la presión arterial, electrolitos séricos y actividad de la renina plasmática. Mientras que las complicaciones del tratamiento con glucocorticoides son infrecuentes (gastritis, insomnio e irritabilidad), las del mineralocorticoide pueden ser más frecuentes. Los pacientes pueden presentar hipokalemia, edema, hipertensión arterial, cardiomegalia e insuficiencia cardíaca congestiva.
Es importante recordar que el uso crónico de esteroides ocasiona un aumento de la resorción ósea y, por tanto, se eleva el riesgo de osteopenia y osteoporosis. En pacientes con insuficiencia suprarrenal secundaria puede requerirse la sustitución de otras hormonas. Se deberá elevar la dosis de glucocorticoides, tanto en la primaria como en la secundaria, previo a cirugías, extracciones dentales o traumatismos importantes. La sustitución de sal y aumento de la fludrocortisona, en la insuficiencia primaria, pueden ser necesarios ante ejercicio intenso, diaforesis profusa, época de calor o pérdidas gastrointestinales importantes.
Insuficiencia Suprarrenal por Supresión Farmacológica
El tratamiento prolongado a dosis altas de corticoides puede provocar insuficiencia suprarrenal debido a supresión sostenida del eje hipotálamo-hipófisis-suprarrenal. Esto se puede prevenir mediante la retirada progresiva y lenta de los esteroides, así como un tratamiento en días alternos hasta alcanzar una dosis equivalente a la de la sustitución habitual. Estos pacientes recuperan la función del eje tarde o temprano, siendo de utilidad la prueba de estimulación con ACTH para confirmarlo.
Tratamiento de la Crisis Suprarrenal
Se realiza la restitución de los niveles circulantes de glucocorticoides y tratamiento de la enfermedad desencadenante, así como del déficit de sodio y agua. Se comienza con 100 mg. de hidrocortisona en bolo IV, continuando con infusión continua de 10 mg/hr o bolos de 100 mg c. 6 a 8 hrs. IM o IV. La restitución de sodio y agua es mediante solución salina al 0.9% o glucosalina. La dosis tan alta de glucocorticoides tiene también un efecto mineralocorticoide, por lo que no es necesario sustituir con fludrocortisona. En ocasiones será necesario el tratamiento con vasoconstrictores.
Referencias Bibligráficas
Cárdenas-roldán J, Rojas-villarraga A, Anaya JM. How do autoimmune diseases cluster in families? A systematic review and meta-analysis. BMC Med. 2013;11:73.
Evangelia Charmandari, Nicolas C Nicolaides. Adrenal insufficiency. The Lancet Volume 383, Issue 9935, 21–27 June 2014, Pages 2152–2167
André Lacroix, Richard A Feelders. Cushing’s syndrome.




